Pairwise Alignment
To align two sequences, navigate to the Pairwise Alignment tab in the Options Panel:
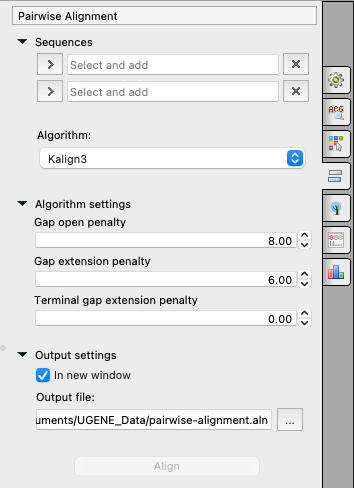
Select two sequences from the original alignment, adjust the parameters, and click on the Align button. The following parameters are available:
Sequences
The first and second sequences for the Pairwise Alignment.
Algorithm
The algorithm for the Pairwise Alignment. There are two algorithms:
- KAlign - provided by the Kalign tool (integrated as an External Tool, check the Data Analysis Tools page for details). This tool uses the Wu-Manber string-matching algorithm. The algorithm details are described in the corresponding publication [Lassmann T, Sonnhammer EL. Kalign—an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics. 2005 Dec 12;6:298. doi: 10.1186/1471-2105-6-298. PMID: 16343337; PMCID: PMC1325270 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1325270/]. The algorithm has the following parameters:
- Gap open penalty - indicates the penalty applied for opening a gap. The penalty must be negative.
- Gap extension penalty - indicates the penalty applied for extending a gap.
- Terminal gap penalty - the penalty for extending gaps from the N/C terminal of protein or 5’/3’ terminal of nucleotide sequences.
- Smith-Waterman - the same algorithm used for the Smith-Waterman Search (check the page for algorithm details - the alignment process works the same way as the searching process). The following parameters are available:
- Algorithm version - version of the algorithm implementation. Non-classic versions produce the same results as classic but much faster. To use these optimizations, your system must support SSE2.
- Scoring matrix - the scoring matrix.
- Gap open penalty - the penalty for opening a gap.
- Gap extension penalty - the penalty for extending a gap.
Output settings
Settings for the output file:
- In the new window - create a new alignment and open it if checked, or just align two sequences of the current alignment if it is not.
- Output file - the path to the result file.