Getting Information About Read
A read displayed in the Reads Area consists of the bases (A, C, G, T). It may also contain the character N, which stands for an ambiguous base. Depending on the value of the Cigar parameter, the read can be shown partially or gaps can be inserted within the read (see below).
By default, when a read is hovered over in the Reads Area, a hint appears:
To disable this behavior, click the following button on the toolbar:

Or uncheck the Show pop-up hint check box on the Assembly Browser Settings tab of the Options Panel.
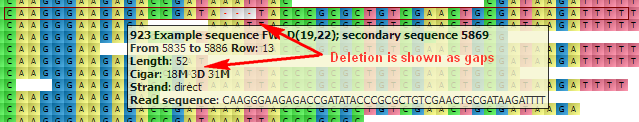
The hint shows the following information about the read:
- Read name
- Location
- Length
- Cigar
- Strand
- Read sequence
The operations in the Cigar parameter are described as follows:
M — Alignment match (can be a sequence match or mismatch).
I — Insertion to the reference. Skipped when the read is aligned to the reference, i.e., it is not shown in the Reads Area but is present in the read sequence.
D — Deletion from the reference. Gaps are inserted to the read when the read is aligned to the reference. For example:

N — Skipped region from the reference. Behaves like D, but has a different biological meaning: for mRNA-to-genome alignment, it represents an intron.
S — Soft clipping (clipped sequences are present in the read sequence, i.e., behaves like I).
H — Hard clipping (clipped sequences are not present in the read sequence).
P — Padding (silent deletion from padded reference).
= — Exact match to the reference.
X — Reference sequence mismatch.
To copy the information about the read to the clipboard, select the Copy read information to clipboard item in the Reads Area context menu. You can now paste it into any text editor.
To copy the current position of the read, select the Copy current position to clipboard item in the Reads Area context menu.