mfold
The tool was added in version 50.
The ‘mfold’ software is a tool for nucleic acid folding and hybridization prediction. It was developed by M. Zuker in the late 1980s for RNA folding and improved for DNA folding in 1996. mfold uses nearest neighbor energy rules.
Tool home page: mfold.org. Similar results to those produced by UGENE can be obtained using the mfold web server (DNA prediction, RNA prediction) or using the OligoAnalyzer tool. For complete documentation on the tool, see a) the website, b) the article, c) the source code in the corresponding folder.
UGENE uses Ghostscript to convert PS files to PNG and PDF.
ToC
- ToC
- Prerequisites
- Open the dialog
- Input parameters
- Saving/resetting dialog state
- Output data
- Tool comparison
- References
Prerequisites
For mfold to work correctly, the path to the temporary directory must not contain spaces, non-printable characters, or non-ASCII characters (Unicode characters such as emoji, Cyrillic, and Chinese family scripts, etc.). In this case, you need to change the path to the temporary directory in the appropriate setting.
UGENE itself must be installed to a path that does not contain non-printable characters or non-ASCII characters (Unicode characters). If installed incorrectly, an error while running the mfold task will be displayed.
Open the dialog
mfold only works with DNA or RNA sequences. You can trigger the dialog form by selecting the appropriate Sequence View Global action  or through the Main Menu→Actions→Analyze
or through the Main Menu→Actions→Analyze  or through the Sequence Context Menu→Analyze
or through the Sequence Context Menu→Analyze 
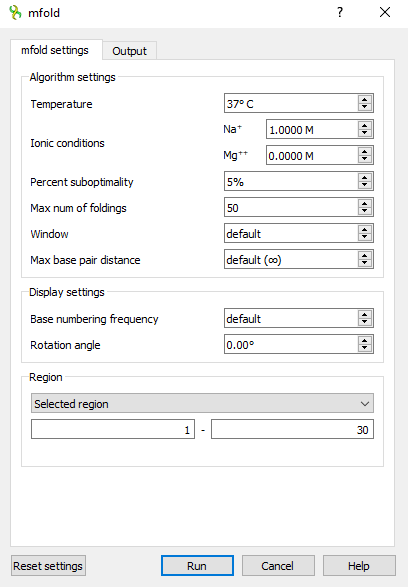
This is what the mfold dialog looks like on Windows OS
Input parameters
The tool is passed explicit and implicit parameters. Explicit parameters are set in the dialog, and implicit parameters are taken from the sequence context.
Algorithm settings
For complete settings details, see the documentation on the mfold website or documentation in source.
This is the Settings section of the mfold settings tab.
| Parameter | Unit | Default | Limits | Tool argument name | Description |
|---|---|---|---|---|---|
| Temperature | °C | 37 | [0,100] | T | The folding temperature. For RNA this is always the default value. For DNA it is taken from the dialog. |
| Ionic conditions | M | Na=1 | [0,1.5] | NA_CONC | Ionic conditions are used to enter total monovalent (Na) and divalent (Mg) ion concentrations. For RNA this is always the default value. For DNA it is taken from the dialog. |
| Mg=0 | MG_CONC | ||||
| Percent suboptimality | % | 5 | [1,100] | P | The suboptimality percentage controls the free energy increment δG for computing suboptimal foldings. Only foldings with a free energy ≤ΔG+δG will be computed, where ΔG is the predicted minimum free energy. Normally, δG=(P/100) |
| Max num of foldings | 50 | [1,100] | MAX | This is the maximum number of foldings that mfold will compute. It is better to limit the number of foldings by careful selection of the P and W parameters. | |
| Window | 0 (len≤29) | [0,50] | W | The window parameter, W, controls the number of foldings that are computed. It may be thought of as a distance parameter. The distance between 2 base pairs, ri rj and ri’ rj’, may be defined as max{ | |
| 1 (30≤len≤49) | |||||
| 2 (50≤len≤119) | |||||
| 3 (120≤len≤199) | |||||
| 5 (200≤len≤299) | |||||
| 7 (300≤len≤399) | |||||
| 8 (400≤len≤499) | |||||
| 10 (500≤len≤599) | |||||
| 11 (600≤len≤699) | |||||
| 12 (700≤len≤799) | |||||
| 15 (800≤len≤1199) | |||||
| 20 (1200≤len≤1999) | |||||
| 25 (2000≤len) | |||||
| Max base pair distance | ∞ | [1,∞] | MAXBP | If the maximum distance between paired bases parameter, MAXBP, is specified, then any base pair, ri rj, in a folding must satisfy j-i≤MAXBP for a linear sequence or min{j-i, len+i-j}≤MAXBP, for a circular sequence (len is the length of the selected region of the input sequence). |
Display settings
Corresponds to the Extended settings section on the main tab.
| Parameter | Unit | Default | Limits | Tool argument name | Description |
|---|---|---|---|---|---|
| Base numbering frequency | 10 (len≤50) | [0,1000] | LAB_FR | Each image marks the number of the base (nucleotide) starting from the beginning 5’. Frequency with which this number will be displayed on an image depends on this parameter. If it is 0, then the number will not appear anywhere. |
Region settings
Specifies the part of the sequence that will be analyzed. The tool will only work with this piece of the sequence in isolation from the whole sequence.
You can select the entire sequence, a custom region, or a selected region if there was one. The region must be less than 3000 bases. It is acceptable to select a small region (<10bp), but there will almost certainly be no folding there.
If several regions have been selected in a sequence (using the GenBank format), then the dialog will only accept the first of them. The remaining selected regions are ignored. But there is an exception: if the sequence is marked circular and the region passing through the end/beginning is selected, then such a region is considered as a whole region and mfold will be launched on it as on an ordinary piece of the sequence (2 parts of the region – one that goes to the end and another that starts from the beginning of the sequence – will be combined into one sequence and analyzed). This is the only case where the start of a region can be greater than the end:
A linear sequence is marked with the symbol 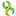 and for it the region 70..50 is invalid. A circular sequence is marked with the symbol
and for it the region 70..50 is invalid. A circular sequence is marked with the symbol  and for it the region 70..50 is valid.
The length of the region in this case 70..50 depends on the sequence length.
and for it the region 70..50 is valid.
The length of the region in this case 70..50 depends on the sequence length.
Output settings
UGENE & Ghostscript settings in the “Output” tab.
| Parameter | Default | Description |
|---|---|---|
| Save output to | /path/to/sequence/ | The folder where the “mfold” subdirectory will be created with the output data in it. By default, this folder is the same as the folder where the input sequence is stored. The output folder must have write permissions. |
| DPI | 96 | Setting up the Ghostscript converter from PS files to PNG. Quality of saved images (PNG files). The higher this parameter, the higher the quality, size, and resolution of the resulting images. |
Internal parameters
Settings that the user cannot explicitly influence.
- Molecule Type (DNA/RNA). mfold gets sequence information from command line arguments. Information about what type will be passed to the tool can be seen next to the sequence name
- Molecule Topology (linear/circular). mfold gets sequence information from command line arguments. If the sequence is marked as circular, then mfold will work with it as a circular one. Otherwise, as with a linear one. Whether the sequence is linear/circular is described here. Part of a circular sequence will also be considered a circular sequence. If you want to change this behavior, uncheck “Mark sequence as circular” and then call the mfold dialog.
- DPI of images in UGENE. Ghostscript has a default DPI of 72. Therefore, this value is used for images for the internal UGENE report and this value cannot be changed.
Saving/resetting dialog state
Some dialog fields retain their state between dialog calls. For such fields, the ability to reset settings is also available (the “Reset settings” button at the bottom left in the screenshot of the dialog). Below is a table showing which states are saved between dialog calls and which of them can be reset to default values.
| Save states | Reset states |
|---|---|
| Algorithm settings | yes (it can be tedious to enter fields every time) |
| Display settings | |
| Region | no (since the region is optionally selected manually by the user) |
| Output settings | yes |
States are saved only for a specific sequence window. Thus, if you have two windows (or two views) for sequence-1 and one window for sequence-2, then each window will have its own state for the mfold dialog.
Output data
There are 3 types of output from the mfold task in UGENE:
- Report displayed inside UGENE.
- HTML report with images saved on disk.
- Temporary data (for advanced users).
UGENE report
Example of a report on Windows (appearance may vary slightly between systems):
- Status, Time, … At the beginning, there is information about the run: information about the task, settings for the launch.
- Output HTML report. Clickable link to the output HTML report. More about this report in the next section.
- Found structures. List of found structures. The structure is characterized by free energy. When clicked, UGENE will scroll the window to the desired structure.
- Structure table. For each found structure, the table contains:
- Thermodynamic details: Free energy, enthalpy, entropy, and an estimated Tm.
- Loop Free-Energy Decomposition: The entire decomposition of the particular folding into loops and stacks, together with their free energies and closing base pairs. Consecutive runs of base pairs are summarized as helices.
- Image. Displays of nucleic acid secondary structure. At the bottom of the image, there is information identifying it: free energy, name of the input sequence, and analyzed region. It has the fixed DPI of 72.
If no structure is found, mfold will fail with an error and display a corresponding message “No hairpins found. Nothing to show”. No new files will be created.
Sometimes the report may not contain the structure table. This happens because there are a lot of results/the resulting decomposition tables are too large. In this case, the result must be viewed in a browser (click on the “Output HTML report” link).
HTML report
A single HTML report and PNG+PDF for each structure found are saved in the output folder. The HTML report displays these images, PDFs, and detailed information about the launch in an easy-to-view form. This version of the report also contains:
- Launch information.
- List of found structures with links to them.
- Information about each structure. The Loop Free-Energy Decomposition is located under the “Show structure detailed info” arrow. The quality of the images in the report depends on the DPI setting in the dialog. When you click on the picture, a PDF file with a structure image opens in a new tab, identical to the picture image, but allows you to zoom without losing quality.
- mfold log.
This report is portable, i.e., if the directory is completely copied (HTML+all PNGs+all PDFs), the copied data can be re-opened in the browser without loss of functionality. Folders can be deleted/renamed; it will not affect the operation of UGENE.
Temporary output
For advanced users.
mfold produces many files. Not all of them are displayed in the reports. However, while the current UGENE session is open, you can see what other files mfold produces.
In the Log corresponding to the [Algorithms][DETAILS] subcategories, you can see the mfold launch command. It depends on where the UGENE temporary directory is set. For example, it might look like this:
“C:\Program Files\Unipro UGENE\tools\mfold\mfold.bat” SEQ=C:\tmp\ugene_tmp\p14120\mfold\d_0\inp.fa RUN_TYPE=html NA=DNA LC=circular T=37 P=5 NA_CONC=1 MG_CONC=0 MAX=100 ROT_ANG=0
In this case, all temporary output files will be located in C:\tmp\ugene_tmp\p14120\mfold\d_0. For information about the purpose and format of this data, please read the article.
Do not modify/move/delete these files. They are read-only.
Tool comparison
As mentioned earlier, the mfold tool can also be launched from the native site or from the IDT platform.
| UGENE | unafold.org | OligoAnalyzer | |
|---|---|---|---|
| Internet connection | not required | must be (http connection) | must be |
| Privacy | share only what you want | results are visible to everyone | results are visible only to you |
| Storing results | stored as long as needed | are removed over time | stored while the session is open (30 minutes) |
| File output completeness | no energy dot plot | there are all files including energy dot plot | there is an energy dot plot but not some other side files |
| Image quality | vector format is represented by PDF files; quality of raster images (PNG) is configurable | vector format is represented by PDF and PS files; quality of raster images (PNG, JPEG) is configurable | no vector format; raster images aren’t configurable |
| Completeness of algorithm settings | some settings for RNA aren’t taken into account; no constraints | all; constraints can be specified | truncated part; no constraints, have their own settings |
| Completeness of display settings | impossible to specify region’s offset relative to original sequence | offset setting | no offset |
| Max sequence length | 3000 | 2400 | 255 |
See more RNA structure prediction software
References
- Article https://www.ncbi.nlm.nih.gov/pmc/articles/PMC169194/
- mfold home http://www.unafold.org/
- mfold DNA prediction http://www.unafold.org/mfold/applications/dna-folding-form.php
- mfold RNA prediction http://www.unafold.org/mfold/applications/rna-folding-form.php
- mfold sources http://www.unafold.org/mfold/software/download-mfold.php
- mfold settings documentation http://www.unafold.org/mfold/documentation/mfold-documentation.php
- Viewing launch results http://www.unafold.org/cgi-bin/view-folds.cgi
- Forum with mfold authors http://www.unafold.org/forum/
- mfold references http://www.unafold.org/mfold/documentation/mfold-references.php
- Implementation of mfold by IDT company (OligoAnalyzer) https://www.idtdna.com/calc/analyzer