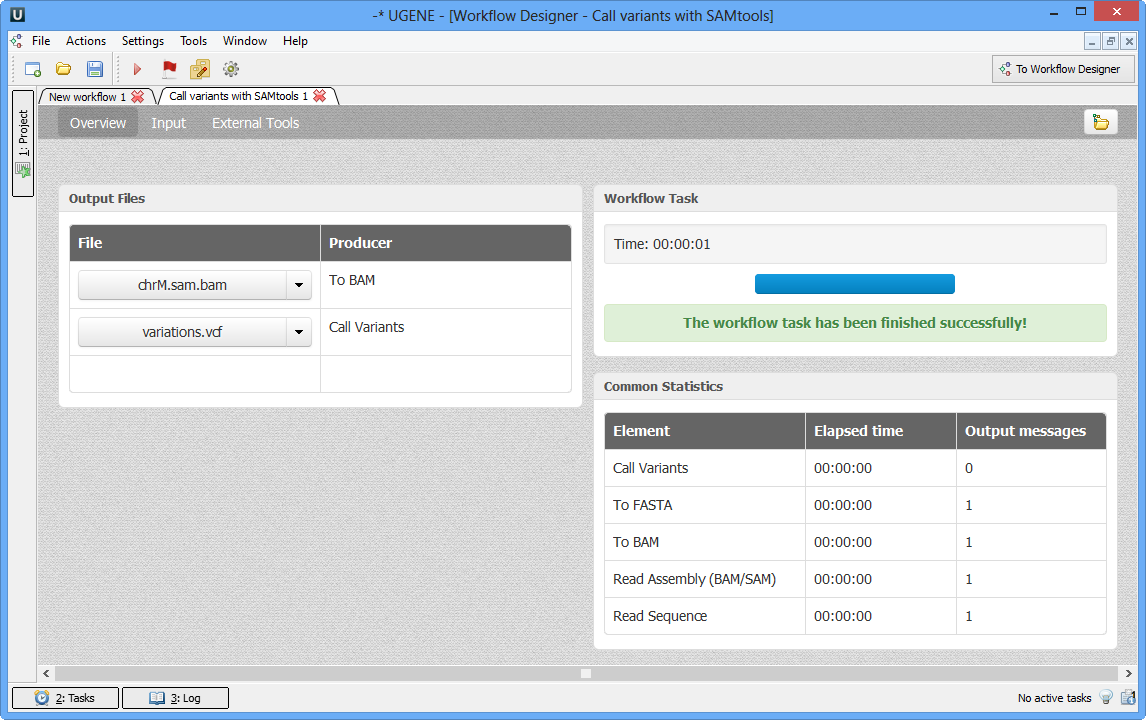
Call Variants with SAMtools
Variant calling in UGENE can be performed using the SAMtools mpileup and bcftools view utilities. For more information about SAMtools and its utilities, visit the SAMTools homepage. Both utilities are embedded into UGENE, so no additional configuration is necessary.
How to Use This Sample
If you haven’t used the workflow samples in UGENE before, please refer to the “How to Use Sample Workflows” section of the documentation.
Workflow Sample Location
The workflow sample “Call Variants with SAMtools” is located in the “NGS” section of the Workflow Designer samples.
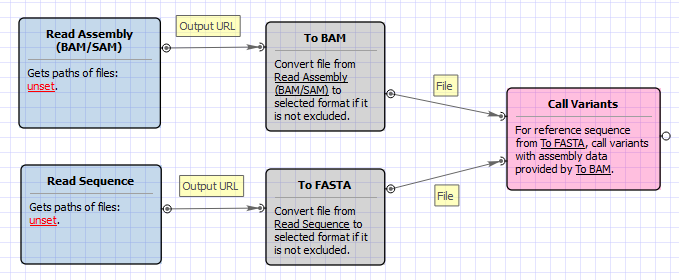
Workflow Image
Workflow Wizard
The wizard consists of 5 pages.
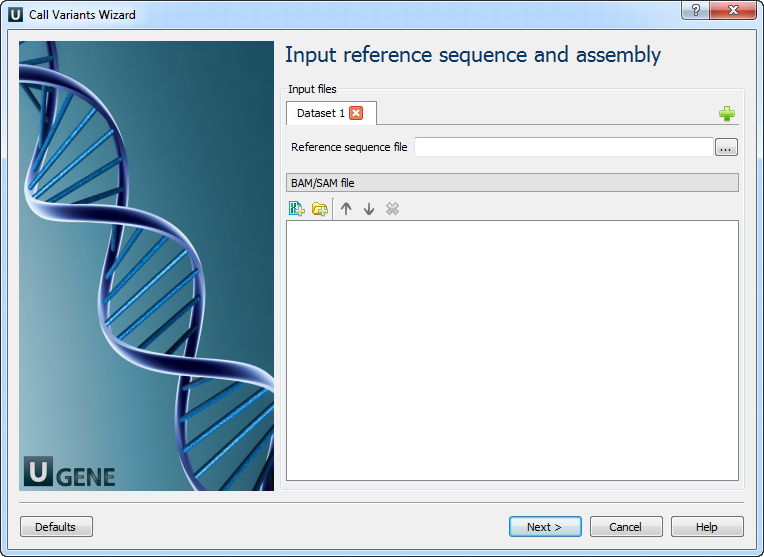
1. Input Reference Sequence and Assembly
Provide a file with a reference sequence and a sorted BAM or SAM file.
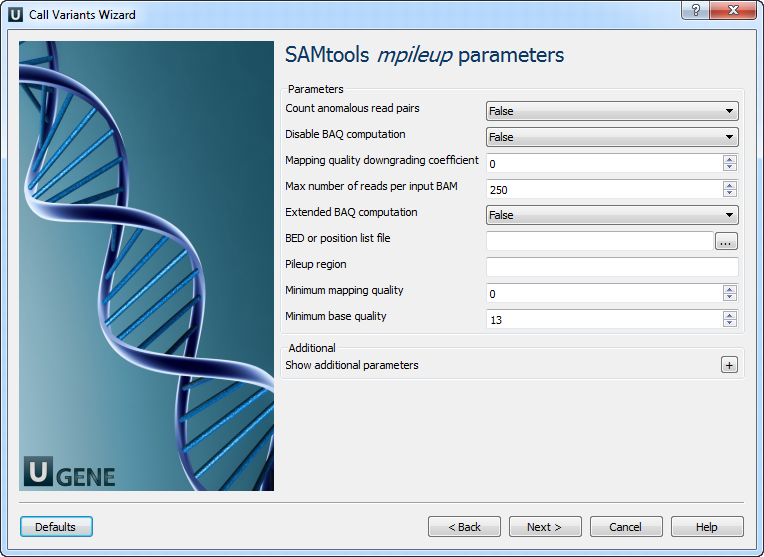
2. SAMtools mpileup Parameters
| Parameter | Description |
|---|---|
| Count anomalous read pairs | Do not skip anomalous read pairs in variant calling. |
| Disable BAQ computation | Disable probabilistic realignment for base alignment quality (BAQ). Helps reduce false SNPs. |
| Mapping quality downgrade | Coefficient for downgrading mapping quality due to excessive mismatches. Recommended: 50 for BWA. |
| Max reads per BAM | Maximum number of reads considered at a position. |
| Extended BAQ computation | Improve sensitivity for MNPs, may reduce specificity. |
| BED or position list file | List of regions or sites to generate pileup. |
| Pileup region | Generate pileup only for the specified region. |
| Minimum mapping quality | Filter alignments below this mapping quality. |
| Minimum base quality | Ignore bases below this quality score. |
| Illumina-1.3+ encoding | Assume quality values are in Illumina 1.3+ format. |
| Gap extension error | Phred-scaled error probability. Lower values allow longer indels. |
| Homopolymer errors coefficient | Used to model indel sequencing errors in homopolymers. |
| No INDELs | Disable INDEL calling. |
| Max INDEL depth | Skip INDEL calling above this per-sample depth. |
| Gap open error | Phred-scaled error probability. Lower values increase indel calls. |
| List of platforms for indels | Comma-separated list of sequencing platforms used for indel calling (e.g., ILLUMINA). |
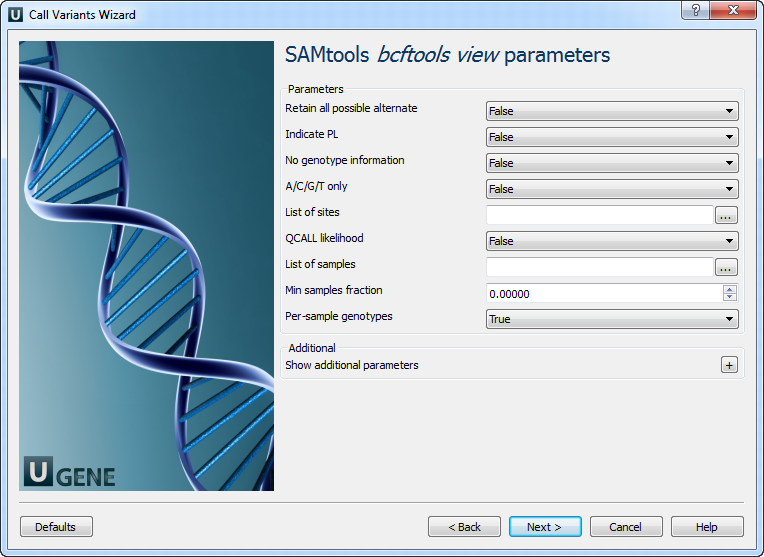
3. SAMtools bcftools view Parameters
| Parameter | Description |
|---|---|
| Retain all alternatives | Keep all alternate alleles at variant sites. |
| Indicate PL | Specify if PL is from older versions (e.g., r921). |
| No genotype information | Do not output per-sample genotype data. |
| A/C/G/T only | Skip variants where REF is not A, C, G, or T. |
| List of sites | Output information only for these specific sites. |
| QCALL likelihood | Output QCALL likelihood format. |
| List of samples | Use a file to specify samples and ploidy. Ploidy can be 1 or 2. |
| Min samples fraction | Skip loci with coverage in fewer than specified fraction of samples. |
| Per-sample genotypes | Enable per-sample genotype calling. |
| INDEL-to-SNP ratio | Ratio of INDEL-to-SNP mutation rate. |
| Gap open error | Error probability for opening gaps (affects indels). |
| **Max P(ref | D)** |
| Pair/trio calling | Enable family-based variant calling. Requires trio configuration (-s). |
| N group-1 samples | Number of samples in group 1 for contrast SNP calling. Outputs: PC2, PCHI2, QCHI2. |
| N permutations | Number of permutations for association test. |
| Max P(chi²) | Perform permutations only for loci with P(chi²) below this threshold. |
4. SAMtools vcfutils varFilter Parameters
| Parameter | Description |
|---|---|
| Log filtered | Print filtered variants into the log. |
| Minimum RMS quality | Minimum root-mean-square mapping quality. |
| Minimum read depth | Minimum read depth. |
| Maximum read depth | Maximum read depth. |
| Alternate bases | Minimum number of alternate bases. |
| Gap size | Filter SNPs near gaps within given distance. |
| Window size | Window size for adjacent gap filtering. |
| Strand bias | Minimum P-value for strand bias. |
| BaseQ bias | Minimum P-value for base quality bias. |
| MapQ bias | Minimum P-value for mapping quality bias. |
| End distance bias | Minimum P-value for distance-to-end bias. |
| HWE | Minimum P-value for Hardy-Weinberg equilibrium (plus F<0). |
5. Output Variations
Configure how the output is written.
The work on this pipeline was supported by grant RUB1-31097-NO-12 from NIAID.