ChIP-Seq Coverage
This workflow sample prepares ChIP-Seq processed data (using BedTools and bedGraphToBigWig) for visualization in a genome browser. Given a BED file as input, it produces a BigWig file.
How to Use This Sample
If you haven’t used workflow samples in UGENE before, check the section: “How to Use Sample Workflows”
Workflow Sample Location
The sample “ChIP-Seq Coverage” is available in the “NGS” section of the Workflow Designer.
Workflow Image
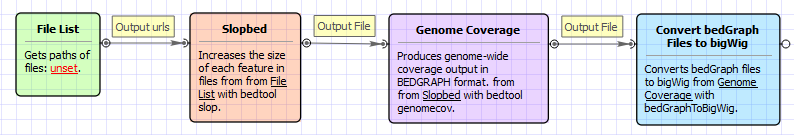
The opened workflow looks like this:
Workflow Wizard
The wizard has 3 pages:
Page 1: Input Data
Upload a BED file with ChIP-Seq tags.
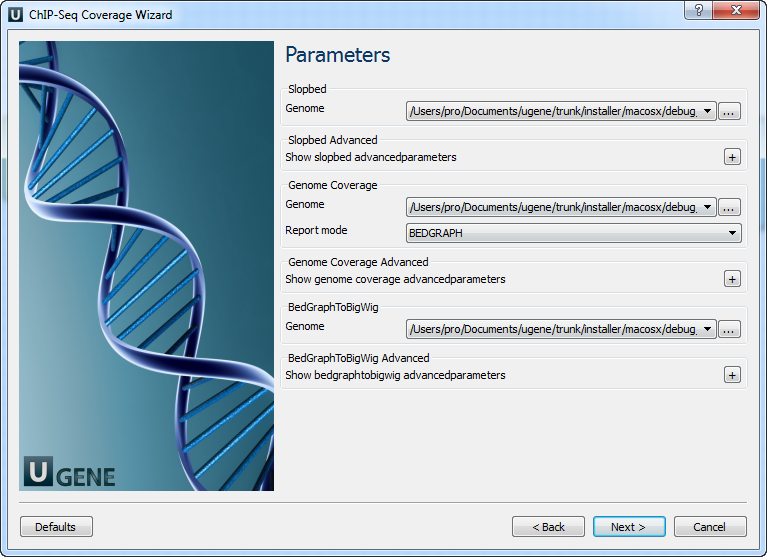
Page 2: Parameters
Modify parameters for SlopBed, GenomeCoverage, and BedGraphToBigWig:
| Parameter | Description |
|---|---|
| Genome | Required by bedtools slop. A genome file that defines the chromosome lengths. (-g) |
| Each direction increase | Extend intervals by N bp in both directions. Overrides -l and -r. (-b) |
| Subtract from start | Subtract N bp from start. (-l) |
| Add to end | Add N bp to end. (-r) |
| Strand-based | Interpret -l and -r based on strand. (-s) |
| As fraction | Interpret -l and -r as a fraction of feature length. (-pct) |
| Print header | Include header from input. (-header) |
| Filter start > end fields | Remove lines with start > end. |
| Report mode | One of: Histogram, Per-base (-dz), Per-base (1-based) (-d), BEDGRAPH (-bg), BEDGRAPH incl. uncovered (-bga) |
| Split | Treat BAM or BED12 entries as blocks. (-split) |
| Strand | Restrict analysis to a strand. Requires strand info in column 6. (-strand) |
| 5 prime | Use only 5’ positions. (-5) |
| 3 prime | Use only 3’ positions. (-3) |
| Max | Combine depths ≥ max into one bin. (-max) |
| Scale | Multiply coverage by a constant (e.g., for RPM normalization). Default is 1.0. (-scale) |
| Trackline | Add UCSC track line. (-trackline) |
| Trackopts | Additional track line definition options. (-trackopts) |
| Block size | Items per R-tree node. (-blockSize) |
| Items per slot | Data points per slot. (-itemsPerSlot) |
| Uncompressed | Disable compression. (-unc) |
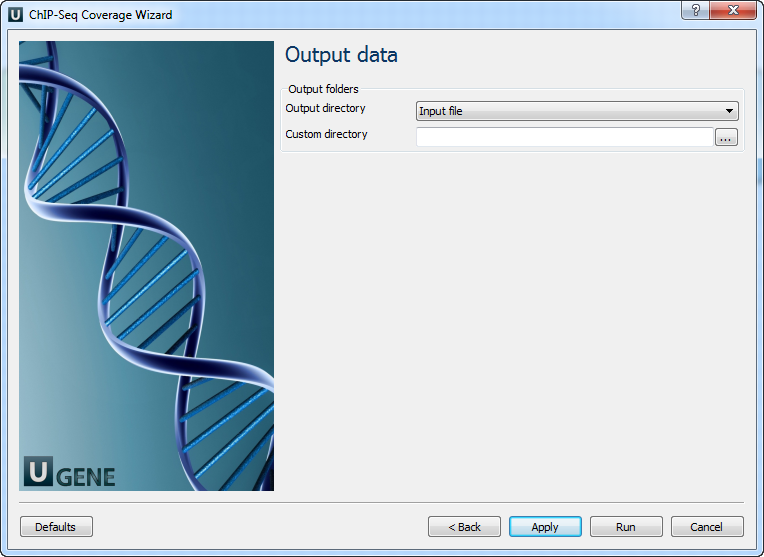
Page 3: Output Files
Select the output directory for the generated BigWig and intermediate files.