De novo Assemble Illumina SE Reads
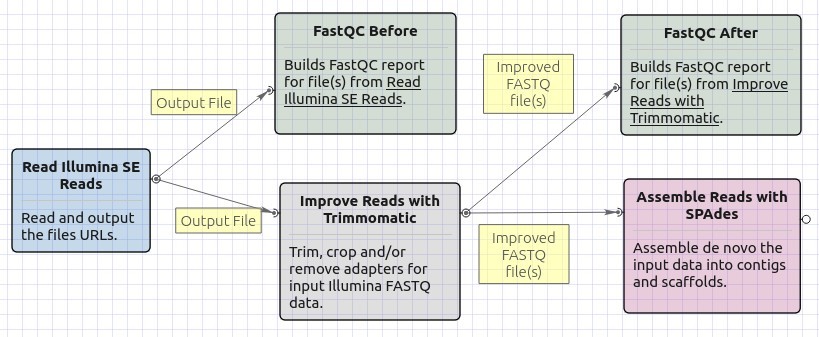
This workflow sample takes FASTQ files with single-end Illumina reads as input and processes them as follows:
- Improves read quality using Trimmomatic
- Generates quality reports using FastQC
- Performs de novo assembly using SPAdes
How to Use This Sample
If you haven’t used workflow samples in UGENE before, check the How to Use Sample Workflows section of the documentation.
Workflow Sample Location
The workflow sample “De novo Assemble Illumina SE Reads” is available in the “NGS” section of the Workflow Designer samples.
Workflow Image
Workflow Wizard
The wizard has 4 pages:
1. Input Data: Illumina Single-End Reads
Specify the input files containing single-end Illumina reads.
2. Trimmomatic Settings
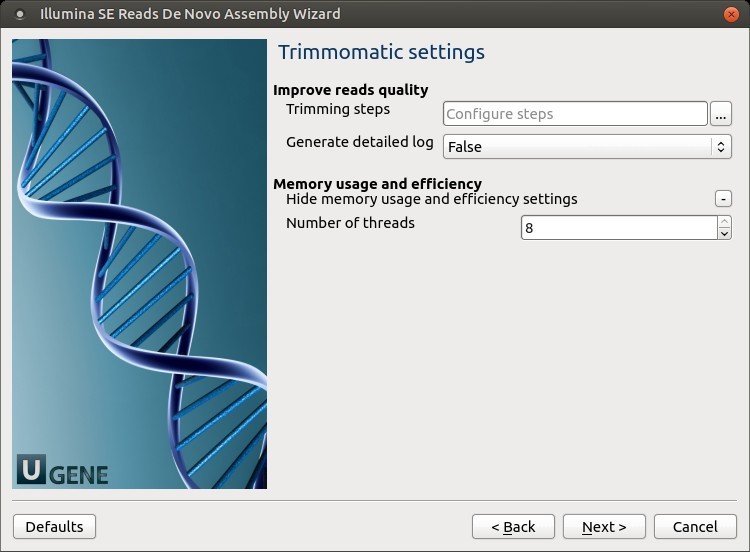
Click the Add new step button to configure trimming steps.
Available trimming steps:
- ILLUMINACLIP
- SLIDINGWINDOW
- LEADING
- TRAILING
- CROP
- HEADCROP
- MINLEN
- AVGQUAL
- MAXINFO
- TOPHRED33
- TOPHRED64
Each step has its own parameters (see the original document for detailed descriptions).
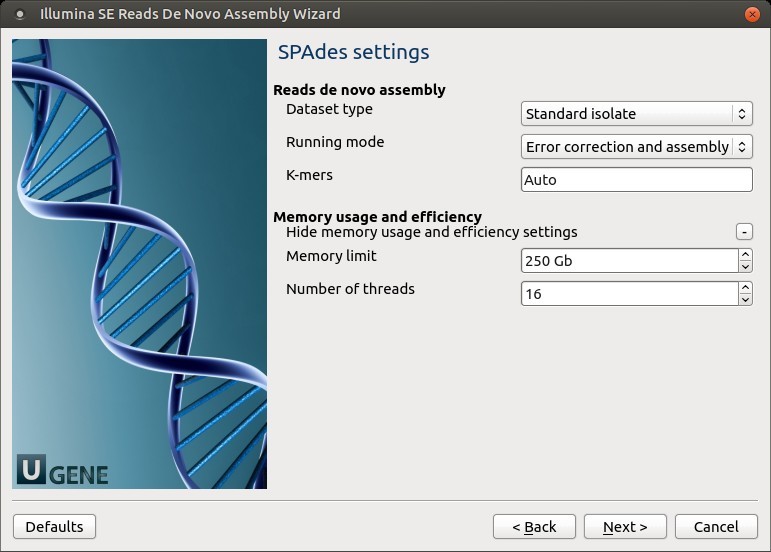
3. SPAdes Settings
Default SPAdes parameters can be customized here.
SPAdes Parameters Table
| Parameter | Description |
|---|---|
| Dataset type | Select the input dataset type: – standard isolate (default)– multiple displacement amplification (--sc) |
| Running mode | By default, SPAdes performs both error correction and assembly. You can select: – --only-assembler (only assembly)– --only-error-correction (only correction) |
| Error correction | Uses BayesHammer for Illumina and IonHammer for IonTorrent data. ⚠️ Avoid if reads lack quality info (e.g., FASTA format) |
| K-mers | Specify k-mer sizes with -k. If omitted, SPAdes selects appropriate values automatically. |
4. Output Files Page
Choose the output directory where results will be saved.