De Novo Assembly and Contigs Classification
⚠️ Attention: The Metagenomics workflow was available before UGENE v40.
This workflow takes FASTQ files with metagenomic NGS reads as input and processes them as follows:
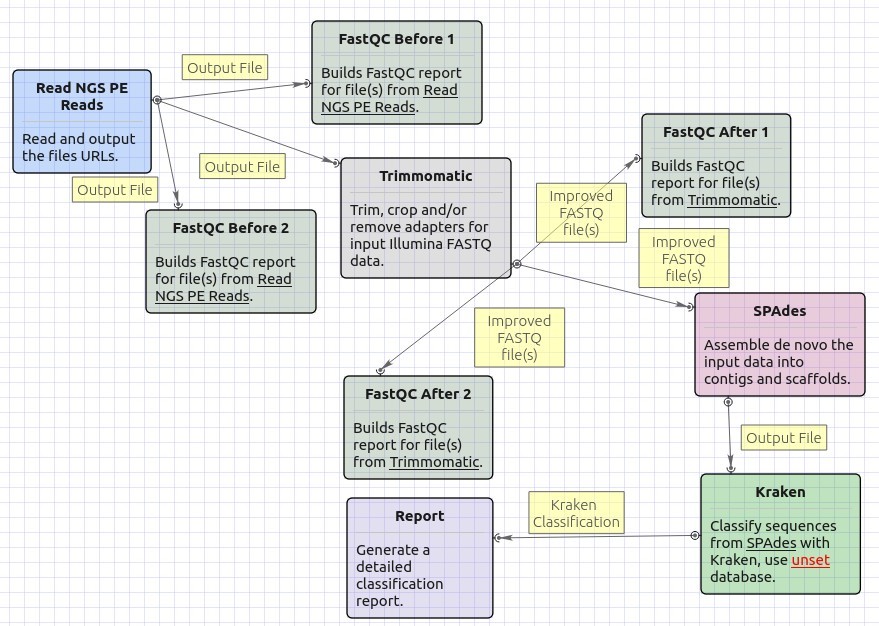
- Improves read quality with Trimmomatic
- Provides quality reports using FastQC
- Performs de novo assembly with SPAdes
- Classifies contigs using Kraken
- Generates a general classification report
How to Use This Sample
If you haven’t used the workflow samples in UGENE before, please refer to the “How to Use Sample Workflows” section of the documentation.
Workflow Sample Location
The sample “De Novo Assembly and Contigs Classification” is in the “NGS” section of the Workflow Designer samples.
Workflow Images
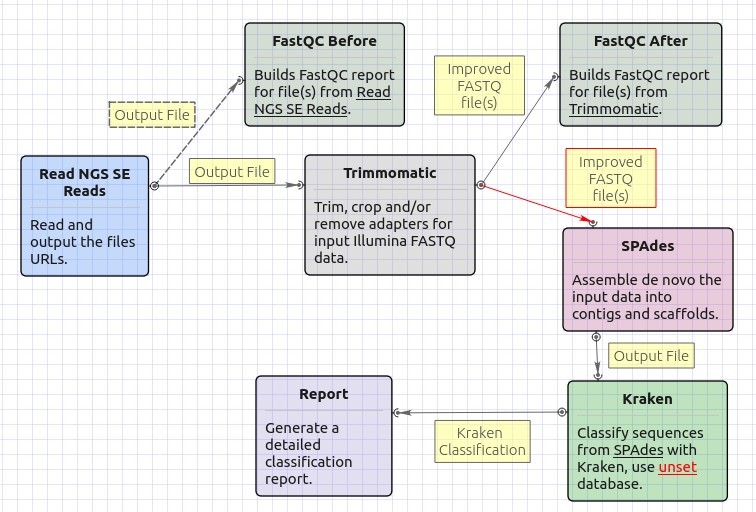
Single-End Reads
Paired-End Reads
Workflow Wizard Overview
The wizard has 5 pages:
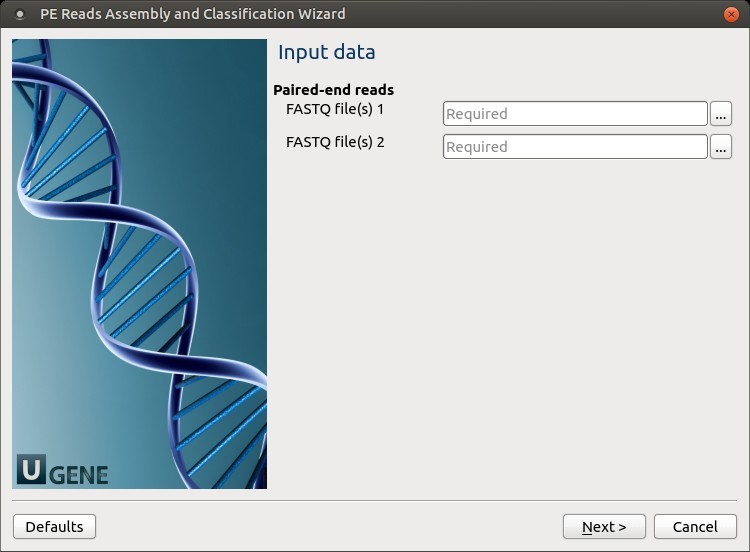
1. Input Data
Set the FASTQ input files.
2. Trimmomatic Settings
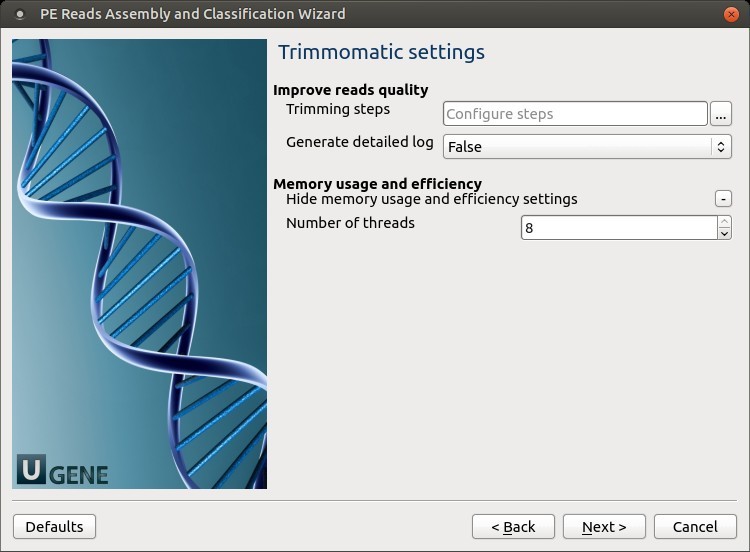
Click the Add new step button to configure trimming steps.
Trimming steps include:
- ILLUMINACLIP
- SLIDINGWINDOW
- LEADING
- TRAILING
- CROP
- HEADCROP
- MINLEN
- AVGQUAL
- MAXINFO
- TOPHRED33
- TOPHRED64
(See original documentation for parameter details.)
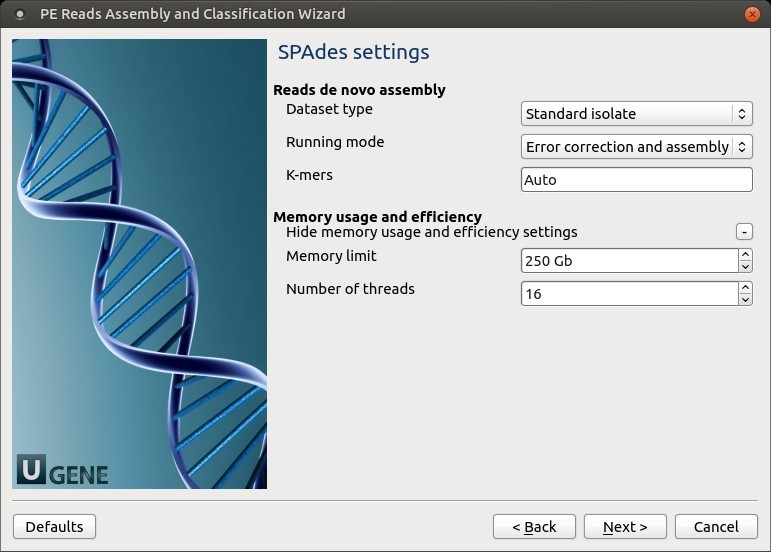
3. SPAdes Settings
You can modify default SPAdes parameters.
SPAdes Parameters
| Parameter | Description |
|---|---|
| Dataset type | Input dataset type: - standard isolate (default)- multiple displacement amplification (--sc) |
| Running mode | Choose mode: - Full (error correction + assembly) - --only-assembler- --only-error-correction |
| Error correction | Uses BayesHammer for Illumina, IonHammer for IonTorrent. ⚠️ Skip for reads without quality (e.g., FASTA). |
| K-mers | Set k-mer sizes using -k. Auto-selected if omitted. |
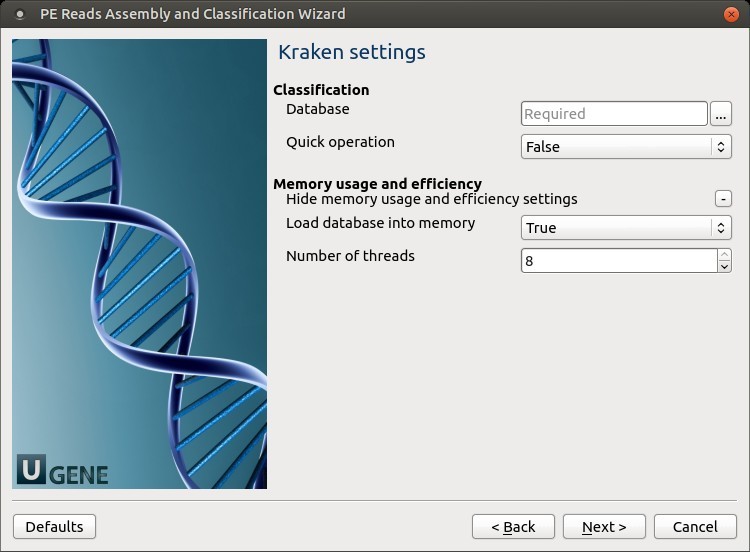
4. Kraken Settings
Configure classification parameters.
Kraken Parameters
| Parameter | Description |
|---|---|
| Database | Path to the Kraken database directory |
| Quick operation | Stop classification early if hit count exceeds threshold |
| Minimum number of hits | Number of hits required before stopping classification |
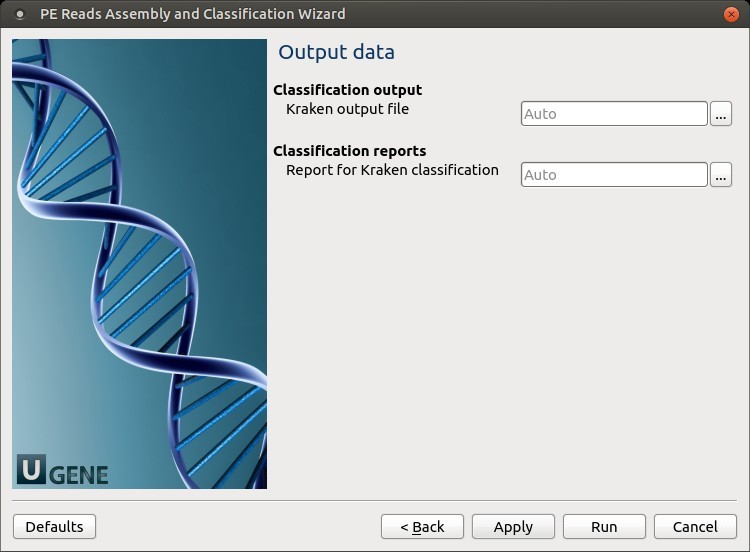
5. Output Files
Choose where to store the output files.