
UGENE Mini-Tutorials: DNA Reverse Complement, Find Repeats and More
How to get a DNA reverse complement sequence
UGENE Workflow Designer is a very effective and convenient system for running calculations with whole datasets, not only with a single sequence or alignment. For example, you can use it for getting DNA reverse complement sequences for a set of input sequences. You can do it with the command line interface of your operating system or with the UGENE Workflow Designer interface.
I. Command line interface:
ugene –task=revcompl –in= –out=
The default output file format is FASTA, but you can change it with the parameter –format (e.g. –format=genbank).
In addition, the parameter –type lets you choose the type of result: reverse complement dna sequence, just reverse ones, or just complement ones. Open the help for the whole list of opportunities:
ugene –help=revcompl
II. UGENE Workflow Designer:
a. Open the file “data/cmdline/revcompl.uwl” from the UGENE installation directory.
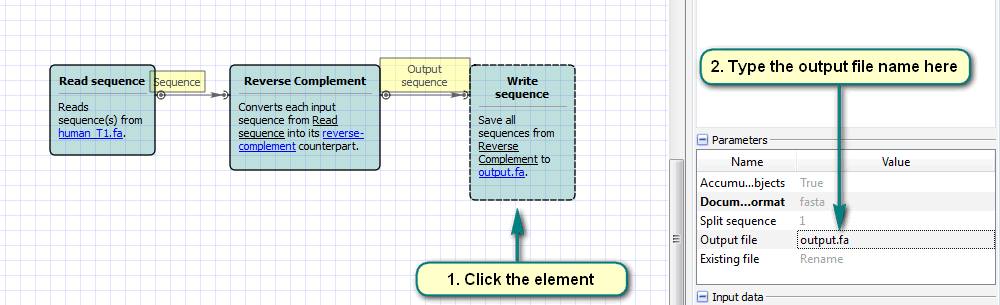
b. Click the “Read Sequence” element and choose the input files.
c. Click the “Write Sequence” element and choose the output file name.
d. Run the workflow.
Here is the documentation page about running this task.
How to find repeats for a set of sequences
If you need to find repeats in each sequence of a set of sequences, you can use UGENE Workflow Designer to automate your work:
1. Open the file “data/cmdline/find-repeats.uwl” from the UGENE installation directory.
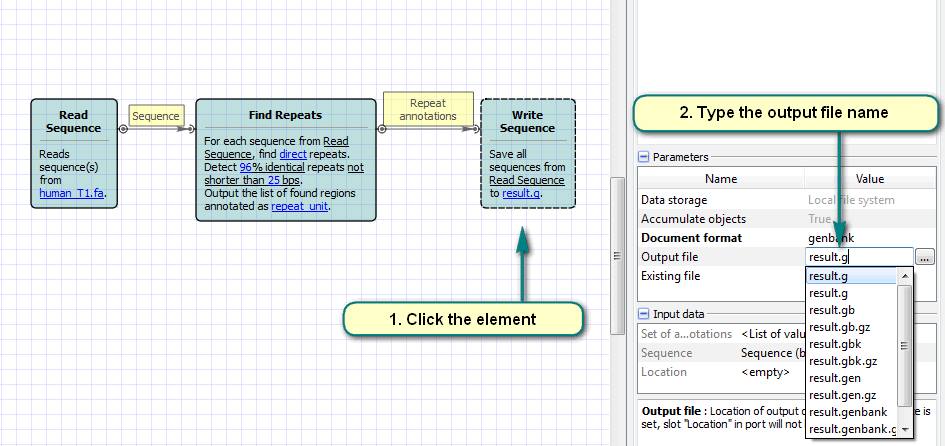
2. Click the “Read Sequence” element and choose files with the input sequences.
3. Click the “Find Repeats” element and set the options of the repeats search algorithm.
4. Click the “Write Sequence” element and choose the output file name.
5. Run the workflow.
The output file will contain all input sequences annotated by the found repeats regions.
The same thing can be done with the command line interface:
ugene –task=find-repeats –in= –out=
Open the help for the whole list of options:
ugene –help=find-repeats
or find them in the corresponding documentation page.
How to create a multi sequence alignment from a set of sequences in a Multi-FASTA file
If you want to create a multi sequence alignment from a set of sequences stored in a Multi-FASTA file, you can use UGENE Workflow Designer and the embedded MUSCLE algorithm:
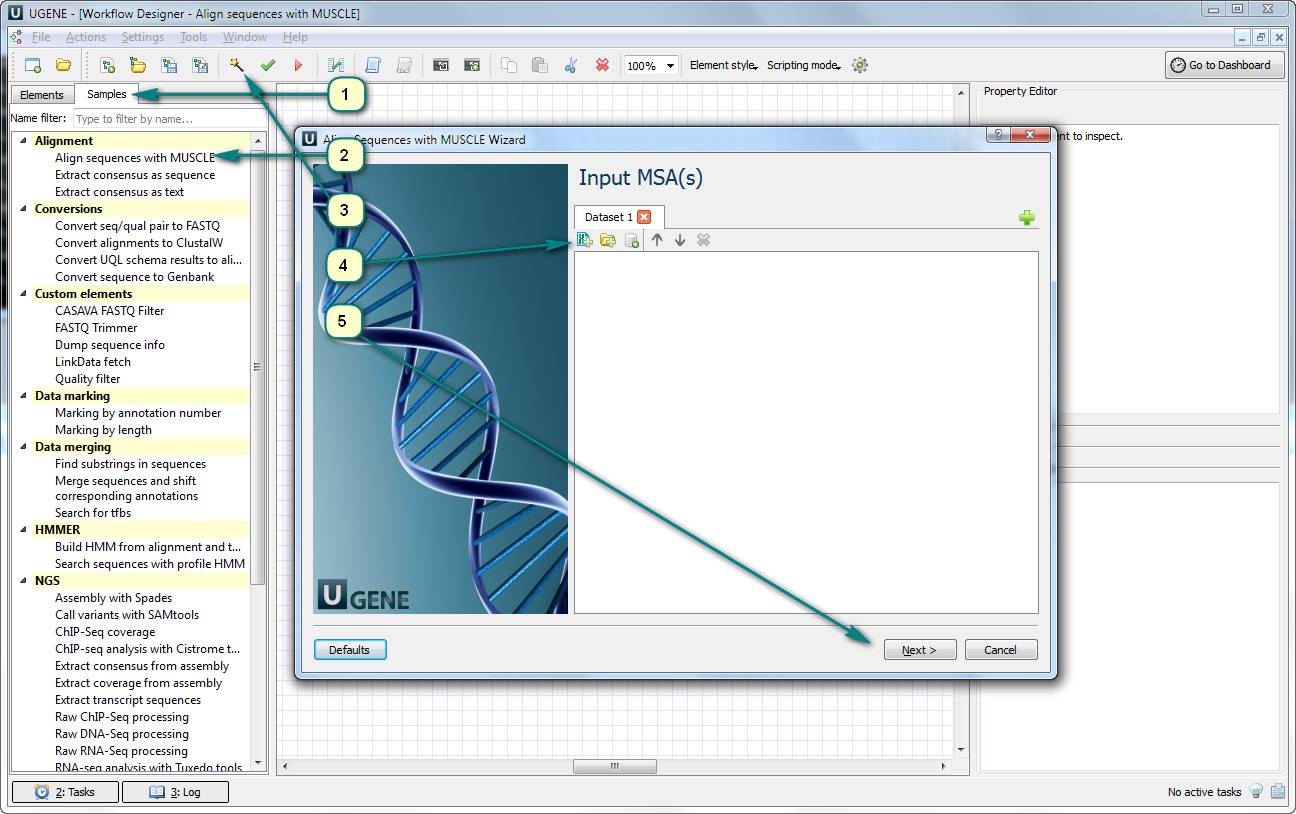
1. Open UGENE Workflow Designer.
2. Click the “Samples” tab and choose «Align sequences with MUSCLE».
3. Click the magic wand button for opening the workflow wizard.
4. Add a file with input sequences. For example, it can be in the Multi-FASTA format – a fasta sequence alignment file.
5. Click “Next” and “Run” buttons.
After the workflow is finished, you will get the result file “muscle_alignment.aln” with the multiple sequence alignment with sequences from the original fasta sequence alignment.
This feature is also available from the UGENE command line interface:
ugene –task=align –in= –out=