
UGENE Mini-Tutorials: SNP Search, SNP Effect and More
UGENE Story
A story about how UGENE Workflow Designer and the team helped a user.
Last week a user sent us an e-mail to ugene@unipro.ru. He tried to solve a task with UGENE and he did not understand how to do it.
The task was the following:
The user had a set of short DNA sequences – the result of Sanger sequencing. Each sequence has the subsequence GCGGCCGCCACC that is the beginning of translation. He wanted to translate each DNA sequence to the protein one starting from that translation beginning and create a multiple alignment from the protein sequences.
If a user of UGENE wants to automate the routine actions on different input data, UGENE Workflow Designer could help with it.
We analyzed the task and decided that UGENE Workflow Designer is really suitable tool for solving it. We created the workflow that:
1) Reads a set of DNA sequences from an input file.
2) Searches for that subsequence in the sequences.
3) Cuts off the part of each sequence before the translation beginning.
4) Translates the cut sequences into the protein ones.
5) Aligns the protein sequences with the MUSCLE algorithm and writes the alignment to a file.
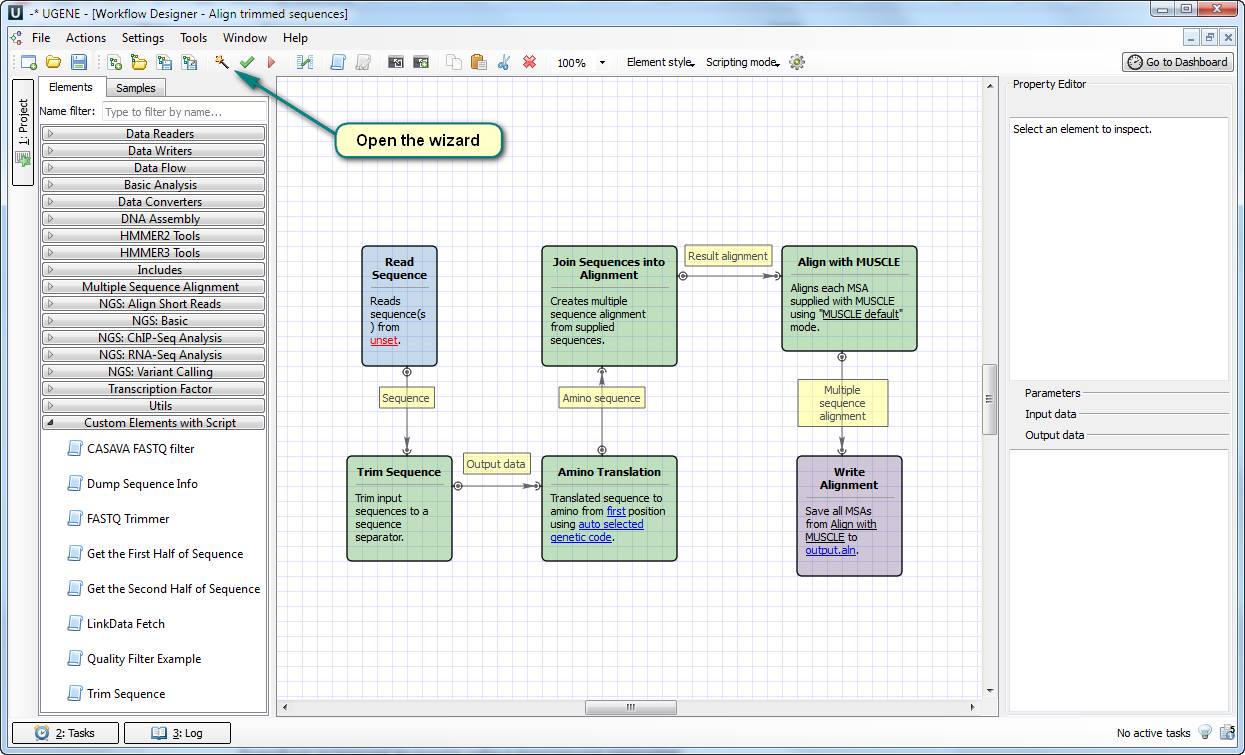
This solution works well for the user because it does exactly the things he wants. You can try the solution by yourself, because all the required files and the test data are attached to this article.
This solution is based on the new element “ Trim Sequence ” for doing such a special translation. You need to add the file “Trim Sequence.usa” to the directory “data/workflow_samples/users/” for making the element available in your UGENE copy. After that, open the workflow file “align trimmed sequences.uwl” and open the settings wizard.
SNP Search
How to do SNP search in NGS data?
The latest piece of news of “How-to in UGENE” (https://www.facebook.com/groups/ugene/permalink/1547052062230702/) has shown you how to get a good alignment from raw NGS data. In this one, you will see how to do the further analysis – to extract a genetic mutation from the alignment.
UGENE Workflow Designer provides a sample workflow that extracts the variations. The workflow is based on the SAMtools utilities.
For running the workflow:
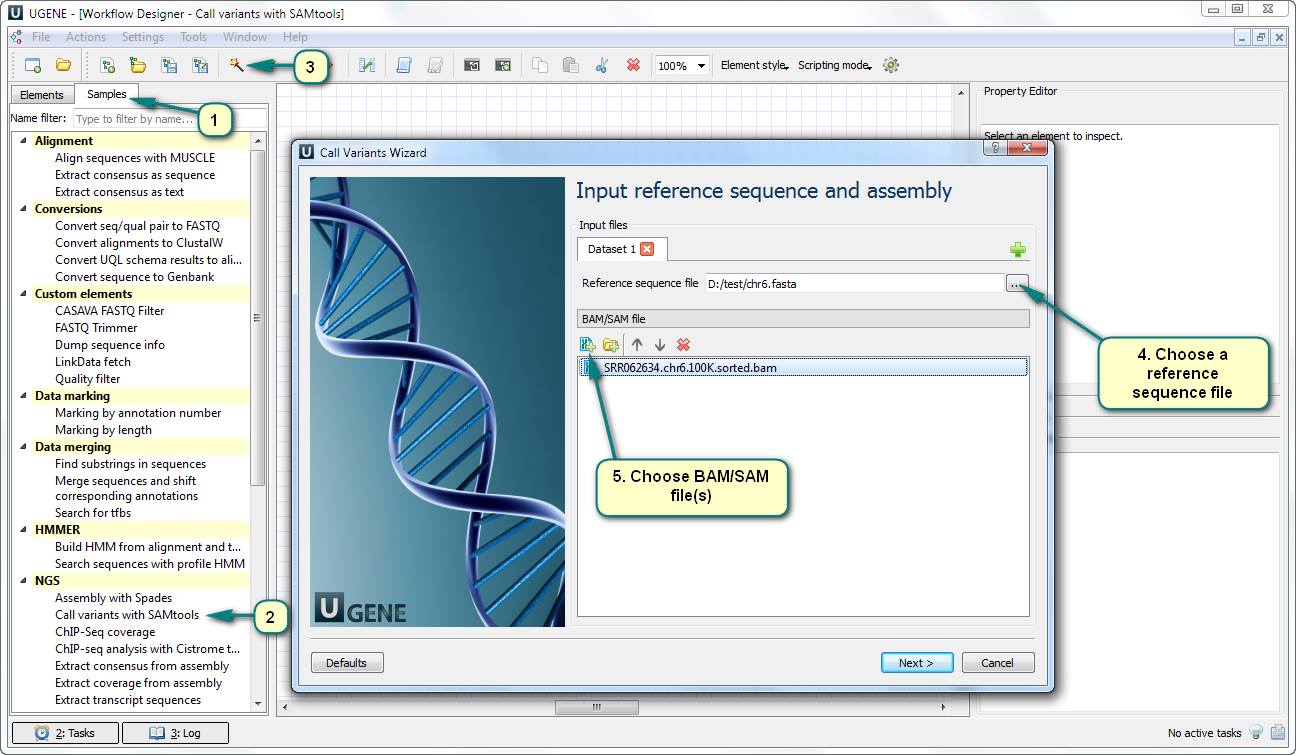
1) Open Workflow Designer and double click the “Call variants with SAMtools” an SNP search sample of the “NGS” category in the “Samples” tab.
2) Open the workflow wizard.
3) Choose a reference sequence file. Usually, this sequence has been used for the mapping (aligning) process.
4) Choose one or several files with alignments. The files must be of the BAM of SAM format.
5) Following the next several of the wizard, you can set up the parameters of the SAMtools utilities (samtools pileup, bcf view, etc.). Let’s just use their default values for the test run.
The result of the workflow is a file of the VCF format. This file contains the extracted genetic mutation.
Follow the updates! In future, we will show you how to do the further analysis of variations with UGENE.
You can find the documentation about this workflow and its parameters here.
A little paper about additional adjusting of the SAMtools utilities (including samtools pileup) is here: http://samtools.sourceforge.net/mpileup.shtml
Sanger Sequencing
Pipeline for automated Sanger sequencing processing
The latest version of UGENE Workflow Designer provides a workflow for processing Sanger sequencing data. The input data of the workflow is a set of files with sequences (reads) and their chromatograms. The workflow:
1) Trims the reads’ ends with the low quality of sequencing.
2) Filters the trimmed reads by minimal length.
3) Aligns the processed reads to a reference sequence.
4) Automatically detects if a sequence of a read should be replaced with reverse-complement one.
The workflow results are:
1) A file with an alignment of the reference sequence and aligned reads.
2) A file with a sequence file (GenBank) annotated by the regions of the aligned reads.
For using the solution:
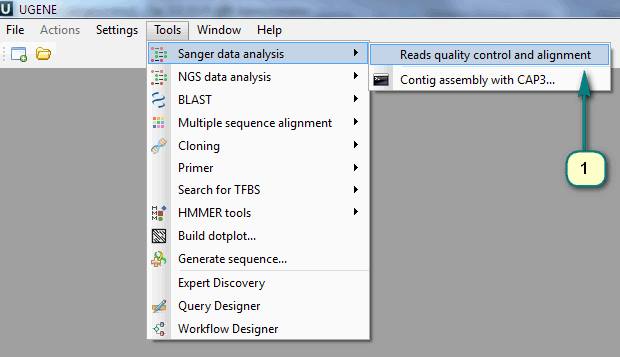
1) Click the menu “Tools -> Sanger data analysis -> Reads quality control and alignment”.
2) Double click the sample description in the appeared Workflow Designer window.
3) Choose the input files: reference sequence and reads.
4) Set the quality threshold and minimal read length.
5) Run the workflow to initialize automated sanger sequencing processing.
The corresponding documentation page.
snpEff
How to annotate variations and predict their effects?
The previous pieces of news of “How-to in UGENE” have shown you how to get a good alignment from raw NGS data (https://www.facebook.com/groups/ugene/permalink/1547052062230702/) and extract variations from the alignment (https://www.facebook.com/groups/ugene/permalink/1553715174897724/). In this one, you will see how to do the further analysis – annotation of variations and effects prediction.
UGENE uses the external program SnpEff for solving this task (http://snpeff.sourceforge.net/SnpEff_manual.html). SnpEff lets you find out the information about variations:
1. Is a variation in a gene, an exon?
2. Does it change the amino-acid of the variation’s position?
3. Does it lead to the premature stop-codon?
4. etc.
For using the variation annotation in UGENE:
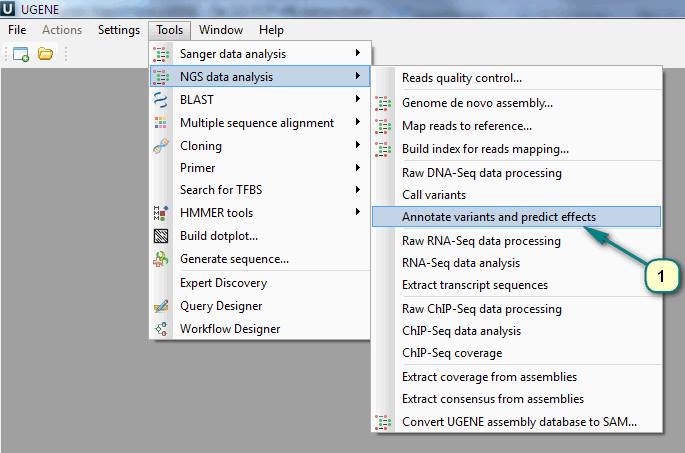
1. Click the menu ”Tools” -> “NGS data analysis” -> “Annotate variants and predict effects”.
2. Double click the workflow preview.
3. Open the workflow wizard.
4. Add the input file with variations.
5. Choose one of the available genomes.
6. Run the workflow and open the report with your web-browser.
Another output file of the workflow is the file with variations (VCF) where each variation is supplemented with the found information.
See the documentation here.