
Tutorial: UGENE as a Transcription Factor Binding Site Prediction Tool
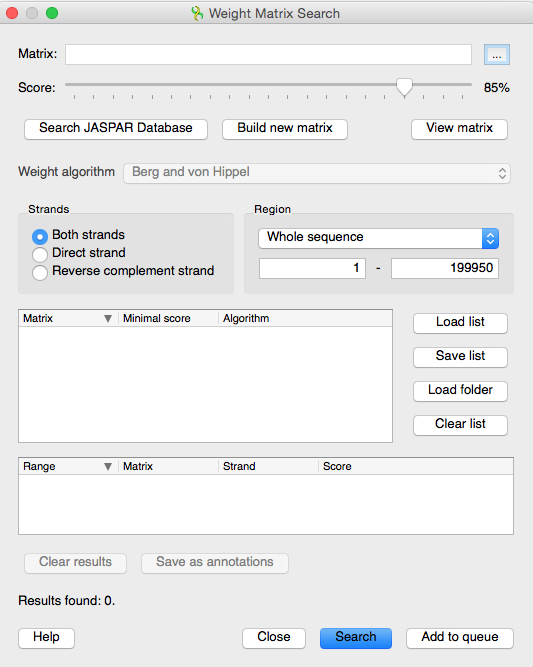
Today's topic is the Weight Matrix plugin, a part of UGENE. It allows to perform the transcription factor binding site search. User can create and save position weight and frequency matrices and align them to an opened sequence. To invoke the plugin, we open a sequence, for the sequence in the human_t1.fa FASTA file and then right-click and select „Analyse“, „Search TBFS with matrices“ to open the transcription factor binding site prediction tool. In the opened dialog box we specify the matrix describing interesting transcriptional factors to search for in the selected sequence, specify the search identity score and the standard transcription factor binding site search parameters such as region and strand.
Transcriptions Factor Binding Site Databases
There're several ways to define the matrix. To start with, UGENE package contains local copies of JASPAR and UNIPROBE databases. By pressing „Browse“ we can select a matrix of one of these databases. Also it is possible to search the JASPAR database with the special interface by pressing „Search JASPAR database“. Here the matrices are divided into categories and we can read detailed information of every matrix represented by the properties. And then we can choose it.
Building Weight Matrix
The last way to choose a weight matrix is to create a specific one from an existing alignment. We press „Build new matrix“ and select certain alignment as the input. The alignment should contain no gaps. After the alignment is set, the Alignment Logo will appear at the bottom part of the dialog box. It gives a representation of the selected alignment.
Running Recognition
Now we need to specify the output file in which the new matrix will be saved And specify the build parameters. Statistic type defines the way in which the statistics will be collected: by subsequences of length one for the mononucleic option or of length two for the dinucleic option. The resulting matrix can be of the „frequency“ type or of the „weight“ type. In the latter case we will need to specify the algorithm to transform intermediate frequency matrix into the resulting weight matrix.
Let's press „Start“. The matrix has been created and saved. Also it has been automatically chosen as the sought-for matrix. Now we can adjust the identity score which stands for the statistical significance of the search. Also the weight algorithm must be set in order to transform the created frequency matrix into the sought-for weight matrix. Let's select the „log-odds“ algorithm. We will search transcriptional factors in both strands of the whole sequence. Press „Search“.
The task is finished and all the transcription factor search results are represented at the table below, including the identity scores. Transcription factor search results can be saved as annotations, which would contain matrix properties if JASPAR or UNIPROBE matrix was used. Let's save the annotations. As we can see, the saved annotations has been automatically added to the opened sequence view, and we can close the dialog and browse the results.