
Release Notes
- Dark theme is now available in UGENE.
- The restriction sites dialog has been visually improved.
- The search field in the restriction sites dialog now supports comma-separated multiple queries.
- Overall stability has been enhanced — more than 20 bugs have been fixed.
- Supported file sharing directly from UGENE for files stored on workspace.ugene.net.
- Added support for the Kraken tool for data analysis.
- Enhanced the color scheme in the multiple alignment editor for better readability.
- Fixed compatibility issues with Linux environments using pre-installed Qt.
- Improved high DPI support on Linux and Windows.
- UGENE Cloud Workspace: workspace.ugene.net
- Enhanced filtering support for restriction sites
- Resolved issues with the external tool, `vcf-consensus`
- Samtools package updated to the latest version
- Added support for restriction sites with multiple cut points
- Fixed a bug in Phylip bootstrap that could lead to incorrect zero distances
- Addressed several crashes in Workflow Designer related to breakpoints
- Improved handling of large datasets that exceed available memory
- Primer3 can be run without a target sequence.
- Primer-BLAST has been implemented in UGENE.
- The hairpin visualization tool mfold has been integrated into UGENE.
- The PlasMapper plasmids annotation database has been updated to the latest version.
- Python, included in the UGENE package, has been upgraded to version 3.
- The list of presets for Primer3 has been expanded.
- Improved compatibility with theme settings in macOS.
- Fixed issues related to the stability and usability of UGENE.
See also a new video about the recent improvements in UGENE.
Sanger files (.ugenedb files with Sanger reads inside) created by UGENE v. 50 will not open in previous versions of UGENE. But there will be no problems when opening "old" Sanger files in the new UGENE v. 50.
- The "Presets" feature has been developed for Primer3, that allows you to select a set of preset settings to run the tool. At the moment, two presets are available - default settings and Recombinase Polymerase Amplification.
- Primer3 has received the function of filtering the resulting primer pairs depending on their homo/heterocomplementarity.
- Annotation selection has been corrected in the Sequence Editor.
- Various minor interface fixes and enhancements.
- Improved macOS support: Dozens of minor interface issues have been fixed.
- Revamped interface for working with restriction sites: New filters and enzyme details have been added.
- Most external tools shipped with UGENE have been updated to their latest versions.
- Enhanced support for PDB and ClustalW file formats.
- Many small issues related to the usability and stability of UGENE on all platforms have been fixed.
- Improved bookmark management. An option to update bookmarks has been added.
- Improved support for large SAM and BAM files.
- The algorithm from the Primer3 tool is now used by default for melting temperature calculations.
- Improved parameter descriptions and functionality in the Primer3 tool.
- Fixed focus issues in several dialogs on macOS.
- The Rebase restriction enzyme database has been updated to the latest version.
- Added the ability to choose commercial suppliers for restriction enzymes.
- Fixed bugs related to annotations and multiline mode in multiple sequence alignment editor.
- A new external tool for building phylogenetic trees: FastTree.
- A new built-in oligonucleotide melting temperature algorithm provided by the Primer3 tool.
- The Phy-tree Viewer has been improved with better scrolling, zooming, preservation of collapse and selection states when switching between different layout types, and more..
- The Assembly Browser has been improved to ensure that the navigational coverage overview does not miss regions for very large references.
- The Msa Editor now supports export of multi-line mode pictures into SVG or regular image formats.
- Improved sequence alphabet detection for extended DNA/RNA alphabets, and enhancements in support for different file formats.
- New visualization mode in MSA Editor: Wrap Mode.
- Phylogenetic tree visualization improvements: hiding nodes, copying of tree image to clipboard, branch curvature.
- Primer3 tool is updated to the latest version.
- Stability and usability fixes.
- Fixed issues with visualization of cladogram and phylogram phy-trees in circular and unrooted modes.
- Improved support for Unicode symbols in file paths for BAM and SAM files used by Samtools module.
- GL2PS library to export vector images of 3D structures was upgraded to the latest version.
- Performance and stability improvements in GFF and ASN format readers.
- MrBayes algorithm now uses the same seed by default and produces repeatable results with the default settings.
- Other stability and usability fixes.
- Assembly browser for a multi-contig reference sequence automatically selects and shows the contig with mapped reads.
- Improved support of MSA alignments with duplicate row names: all phy-tree algorithms integrated into UGENE were fixed.
- MrBayes phy-tree tool integration uses 'random seed' parameter correctly and produces repeatable tree results.
- Zoom-to-selection in MSA Editor fits the selected area into view correctly.
- Dotplot view region selection by mouse clicks was improved.
- Experimental shared MySQL database support is removed from UGENE.
- Many small interface improvements and stability fixes.
- A new widget in MSA Editor: Exclude List.
- Custom algorithm selection support in 'Re-align selected sequences to the alignment' action.
- MSA Overview was optimized and is ON by default for any alignment sizes.
- MSA Editor performance while browsing huge alignment files is improved.
- New dedicated controls for popular options in IQ-TREE dialog.
- Double click on a sequence name in MSA Editor scrolls between start and end of the sequence into the view.
- Fixed order of joined and complemented annotation sequences during the export.
- Create Annotations widget uses standard Genbank feature names by default.
- "Map reads to reference" algorithm is optimized to handle a large count of reads.
- Regression with a new MySQL database connection is fixed.
- Newick file format support is improved.
- A new tool to build phylogenetic trees: IQ-TREE.
- Opening of very large alignments does not block UGENE anymore.
- A double-click in Sequence Viewer does not produce an unexpected selected region.
- Stockholm format parser now supports Unicode correctly.
- FASTA files with a very large number of sequences now can be opened as a multiple sequence alignment.
- ABIF files with incorrect dates do not cause a crash anymore.
- A search by a long list of patterns in Sequence Viewer is not disabled anymore.
- Stability and memory consumption of PCR and Smith-Waterman algorithms are improved.
Alignment Editor
- Multi-row selection support.
- The "Build Tree" dialog now has all PhyML algorithms listed.
- A new option to move selected sequences from one document to another.
- The grouping mode: sorting by the size of a group and showing group sizes in the name list.
Sanger Editor
- A new visualization mode: show alternative mutations.
- Several vertical scrolling bugs have been fixed.
Sequence Viewer
- Speed, memory usage and precision of sequence graphs have been improved.
- Stability problems of auto-annotation routines have been fixed.
- Bugs, which appears while working with sequences larger when 2Gb, have been fixed.
Other improvements
- The "Smith-Waterman" algorithm support for CUDA has been improved.
- The Unicode support has been added for Newick, Plain text, Diff, VCF, SNP file formats.
- The "Random Sequence Generator" memory footprint and generation speed have been improved.
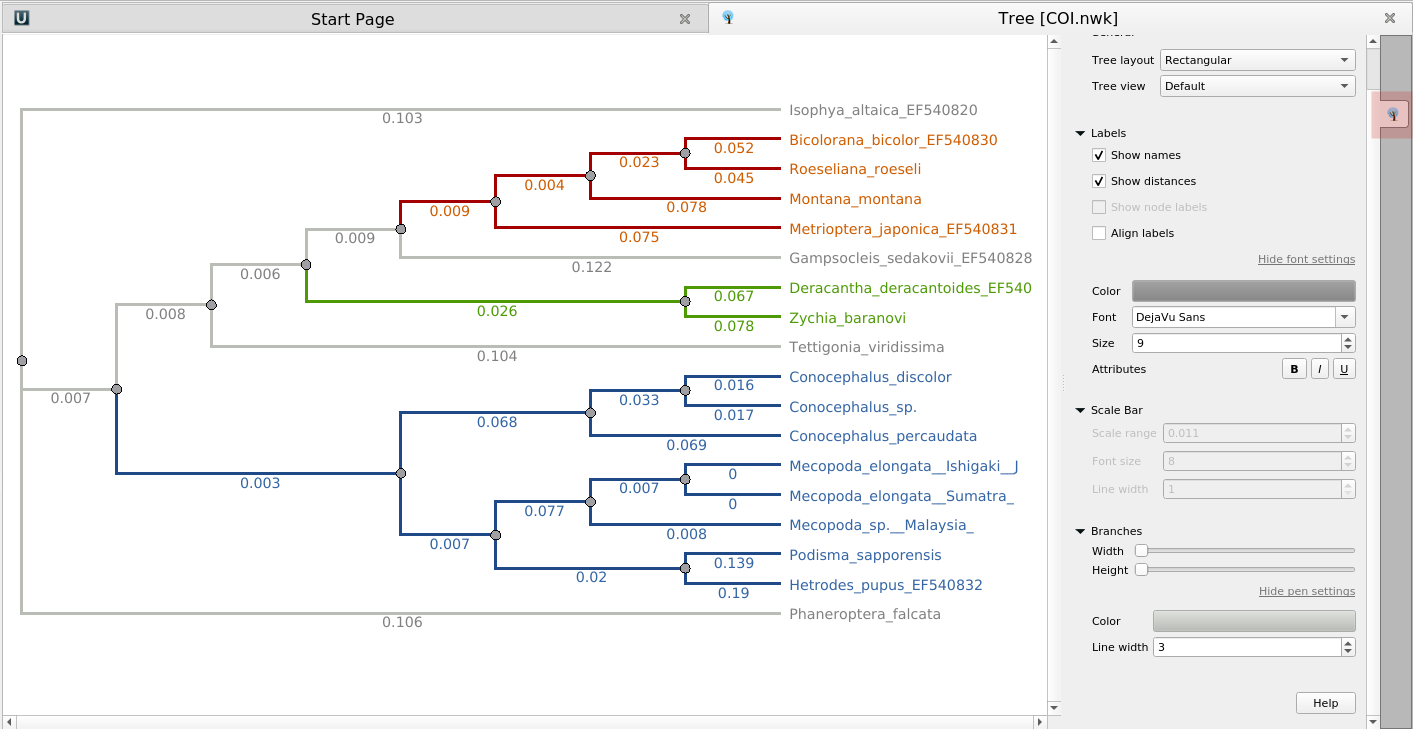
The new version includes the following new features and improvements:
- Phylogenetic tree viewer now allows setting custom colors and fonts for subtree branches and
labels:
- The "Align to alignment" option got support for algorithm selection: ClustalO, MAFFT, Muscle, or
UGENE.:

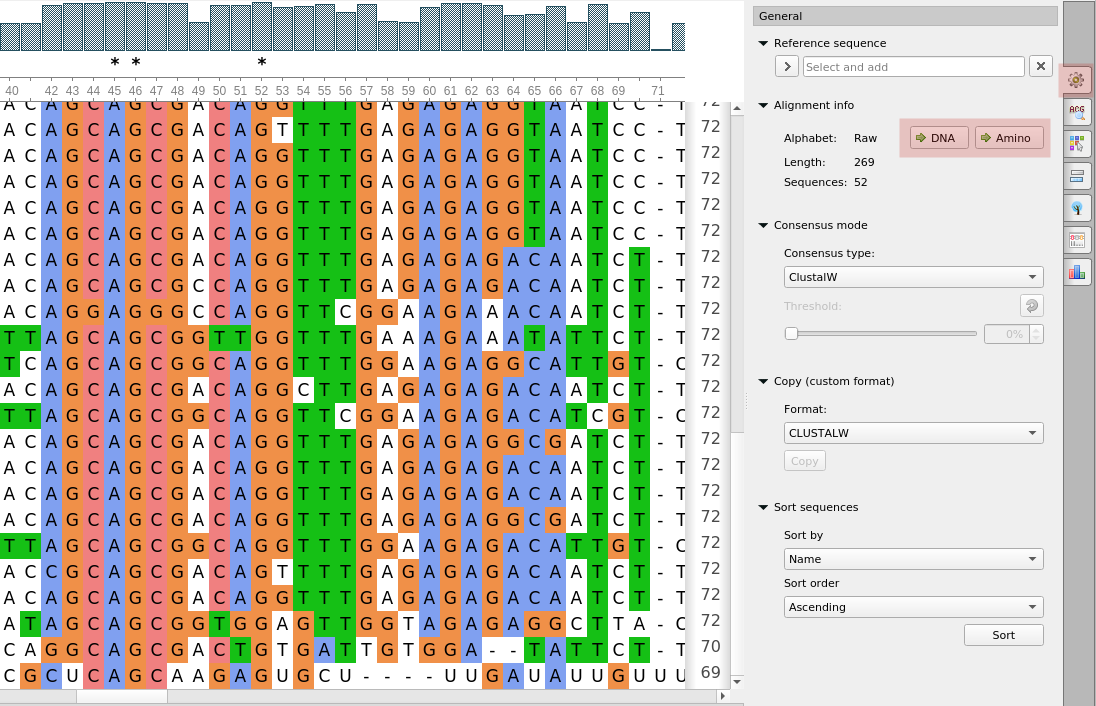
- Alignments with the RAW (undefined) alphabet can be converted to the DNA, RNA, or Amino
alphabets:
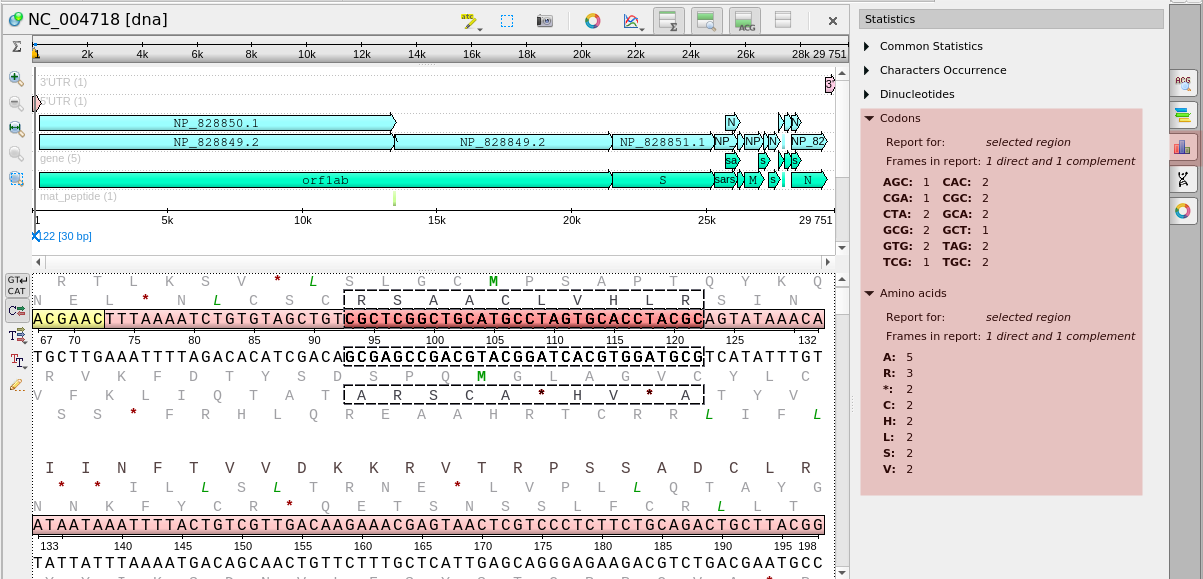
- Sequence Viewer got a new option to show codons or protein statistics for selected regions:
- Other changes:
- Improved Unicode support for FASTA, MSF, and CLUSTALW formats.
- UGENE now speaks Turkish. Use menu Settings/Preferences to update the language.
- Other bug fixes and improvements.
The new version includes the following improvements:
- Export amino acid representation:
The «Export Amino Acid Translation» dialog has been improved. Now it’s possible to choose whether you need to export gaps or not and if yes it’s possible to export them as «X» or «-» symbols. Also, it’s possible to choose the translation frame.
- Sorting features:
«Sort by leading gaps» is available now! Also, all sorting functions work not only for the whole alignment, but for selected reads too.

- The «Cut» function:
Now it’s possible to cut the selected part of the alignment. Find the corresponding item in the context menu or use the following shortcut: «Ctrl+X».

- Other changes:
- Minimum font size has been decreased in Alignment Editor.
- Double-click on a read name moves the screen to the meaningful symbols in Sanger Reads Editor.
- Other bug fixes and improvements.
You may see the full list of improvements here.
- Alphabetical order of External Tools:
Pay attention that the list of the «External Tools» in the «Application Settings» dialog has become sorted alphabetically.
- The copy/paste and naming mechanism in the Alignment Editor has been improved:
The copy/paste function saves the name of the copied sequence now and also roll this name to make it unique. Besides, the possibility to insert sequence to a certain place (near the current selection) has appeared.

- Naming in the «Bookmarks» tab has been improved:
File and object names writing has become more convenient in the «Bookmarks» window.

- Other changes:
- The validation process of External Tools has been improveds.
- Trimmomatic, BWA, and Bowtie2 have been updated.
- A large number of bugs and errors, related to working with the database, external tools, and other scripts, have been fixed.
You may see the full list of improvements here.
The new version includes the following improvements:
- Adding the UGENE launch shortcut on the Desktop:
Pay attention to the appeared «Create desktop shortcut» button in the «Help» menu.
- The «Copy annotations» function is improved in the sequence view:
Copying of selected annotated sequences is tied to the strand of the sequence the current annotation is located on. If there are several annotations on both direct and complementary strands are selected, clicking the «Copy» button causes the copying of sequences from the corresponding strands.

- Other improvements:
- MySQL 8 is supported.
- Phylogenetic trees visualization is improved.
The full list of changes can be found here.
The new version includes a bunch of improvements in Alignment Editor:
-
Sorting sequences in alignment by name and by length: You can sort sequences in alignment
using
the Sort submenu in the Actions main menu or from the context menu.Also, you can use the General Options Panel tab:

There are four ways to sort sequences:
- By name, ascending
- By name, descending
- By length, ascending
- By length, descending
-
Grouping sequences in alignment by sequence names: To search for a pattern(s) in alignment go
to
the “Search in Alignment” tab of the Options Panel.To group results press the Group button. The results will be selected and grouped to the top after the first
click or to the bottom after the second click to the button.
-
Search by sequence names in alignment:
To search by sequence names use the Search in Alignment Options Panel tab with the “Sequence Names” search context.

-
“Weak Similarities” color scheme: When there is a choice between two characters and
it is not
specified by other rules which character to color, following priorities: T>U>G>C>A>B>D>H>K>M>R>S>V>W>Y>N.Coloring rules:
- Do not color gaps (“-“), i.e. use a black font and white background.
- Residues should be colored according to their percentage number in the column (use the
priorities,
described above, in case of equal percentage numbers). - Residues with the most frequency/priority in the column.

-
Other modifications include:
- The ClustalO tool is updated to the latest version.
- “Navigation” and “Appearance” menus in Alignment Editor were restructured for better usability.
- “Edit” menu in Sequence View got a new structure.
The new version includes a lot of useful features in the Alignment Editor (AE):
-
Realigning sequence to other sequence in an alignment: select one or several sequences in an
alignment and click
 to other sequence' button on the AE toolbar") on the AE toolbar. The
selected sequence will be re-aligned to the
other sequences in the alignment.
on the AE toolbar. The
selected sequence will be re-aligned to the
other sequences in the alignment.
-
Advanced search options: a bunch of
features
for searching in an alignment was added: search of several patterns at once, usage of different
algorithms besides the exact pattern match,
search in particular columns. Similarly to the Sequence View, the options are now located on the options
panel tab.

- Appearance of a selected region was improved: the selected region is more colorful, the dashed border is wide.
- Selection of the same region in other sequence: if a region is selected in an alignment and it is required to select the same region in another sequence in the alignment, use the Ctrl (or Cmd on macOS) key and click on the required sequence name.
- Collapsing mode bug-fixing: a few issues with alignments in collapsing mode were fixed.
Modification in the Sequence View (SV) include:
-
State of the SV components is saved: appearance configuration of different SV components is now
saved between different UGENE
sessions.
For example, if you hide the Zoom View or show all translation frames, this will be applied to the next opened SV window even after the program re-start. -
Sequence statistics: the statistics calculation for a nucleotide sequence was corrected, info for
double-stranded molecules was added,
see documentation.

- Copy/paste of annotations: several copy/paste options were added for a selected annotation (similarly to a selected sequence region).
- Circular sequence analysis bug-fixing: a few issues with analysis of a circular sequence were fixed.
Other modifications include:
- New version of the SWISS-PROT format is supported.
- BLAST+ and remote NCBI BLAST are only supported now, the obsolete BLAST was removed.
- The number of UGENE packages was decreased, 32-bit operating systems are no more supported. The old packages are available here.
- The Windows packages are now digitally signed by Unipro.
The new version introduces a bunch of improvements for adding a custom tool into UGENE, in particular:
- Import of your tool into UGENE external tools: the list of external tools is available in the UGENE Application Settings dialog. Here for each tool you can provide a path to the tool executable file. Since UGENE version 33 you can also specify your own tools. See documentation for details.
- Enhanced wizard for creating a workflow element for the tool: to use a custom tool in the UGENE
Workflow Designer graphical interface,
create a workflow element that specifies the tool input/output, parameters, and the command. To start
the wizard click
 on the WD toolbar. See
details here.
on the WD toolbar. See
details here.
- Tighter integration with the WD dashboard: a file or a folder, produced by a parameter of type
“Output file URL” or “Output
folder URL”, automatically appears on the dashboard.

Another improvement in UGENE version 33 is a new color scheme “Percentage Identity (colored)” in the Alignment Editor, see description in the documentation.
' color scheme in the Alignment Editor")
The new version includes an update of the NGS data analysis framework: MetaPhlAn2 tool and other minor improvements.
MetaPhlAn2 (METAgenomic PHyLogenetic ANalysis) is a tool for profiling the composition of microbial communities (bacteria, archaea, eukaryotes, and viruses) from whole-metagenome shotgun sequencing data. The tool relies on ~1M unique clade – specific marker genes identified from ~17,000 reference genomes (~13,500 bacterial and archaeal, ~3,500 viral, and ~110 eukaryotic).
Into the UGENE GUI the tool is integrated as a workflow element. It is included to the “Parallel NGS reads classification” sample workflow:

See also:
- Video about metagenomic workflows in UGENE 1.31-1.32 versions
- Tutorial about metagenomic classification with Kraken
- If you use metagenomic workflows in UGENE please cite this publication.
This is a patch release that fixes a critical issue on macOS Mojave that have recently come out.
Also, the update includes bug fixes for:
- exporting consensus in the Sanger Reads Editor
- downloading a sequence from the Ensembl database
- Quality control: a new tool Trimmomatic was integrated for quality control of NGS reads. It
allows one to cut Illumina adapters, trim
reads ends by quality, trim reads by length, etc.
 The tool is available as a workflow
element and included into several sample workflows (see below, for example).Trimmomatic is a
java application. Install Java
Runtime Environment
and configure it in the UGENE Application Settings to use the tool.
The tool is available as a workflow
element and included into several sample workflows (see below, for example).Trimmomatic is a
java application. Install Java
Runtime Environment
and configure it in the UGENE Application Settings to use the tool.
- De novo assembly: SPAdes was updated to version 3.12.0. Graphical interface for the tool in UGENE
was updated. It is now possible to
input hybrid data, for example, assemble Illumina and Oxford Nanopore reads.Begin with sample workflows
available from “Tools > NGS
data analysis > Reads de novo assembly (with SPAdes)” item in the UGENE main menu.
 Modify the SPAdes input type, if required. SPAdes requires at least one
library of the following types:
Modify the SPAdes input type, if required. SPAdes requires at least one
library of the following types:
- Illumina paired-end/high-quality mate-pairs/unpaired reads
- IonTorrent paired-end/high-quality mate-pairs/unpaired reads
- PacBio CCS reads
Additionally, one can provide PacBio CLR, Oxford Nanopore, Sanger reads or contigs.
See “Input data” parameter of the SPAdes workflow element.
Hint: hold Alt key and use mouse cursor to move a port on a workflow element.
Note that SPAdes is available on macOS and 64-bit Linux only.
- Metagenomics: a new infrastructure for taxonomy classification of whole-genome shotgun sequencing
data was developed. This includes tools
Kraken, CLARK, DIAMOND, WEVOTE and other. Reference data for the tools are also provided: NCBI taxonomy
information; RefSeq data for viruses,
bacteria, human; etc. It is recommended to use the Online Installer UGENE package to install and
configure the data, check required items in the
“NGS additions: metagenomics classification” item:
 Several sample workflows with the
tools are available from “Tools > NGS data analysis > Metagenomics classification”
item in the main menu. In particular, the
following workflow executes Kraken,
CLARK,
and DIAMOND tools in parallel. After that, the results are merged by WEVOTE.
Several sample workflows with the
tools are available from “Tools > NGS data analysis > Metagenomics classification”
item in the main menu. In particular, the
following workflow executes Kraken,
CLARK,
and DIAMOND tools in parallel. After that, the results are merged by WEVOTE. Besides classification, building of a
custom database is supported for each tool
(see, for example, “Build
Kraken Database”
workflow element).Note that the computer should be powerful enough to run the workflows (at least 8Gb of
RAM is required). The whole package
requires about 230-240 Gb of disk space (see details here).
Trimmomatic and FastQC tools, included into the sample workflows, require Java
Runtime Environment to be installed and configured in the UGENE Application Settings. The other
tools are provided for macOS and 64-bit
Linux operating systems only.
Besides classification, building of a
custom database is supported for each tool
(see, for example, “Build
Kraken Database”
workflow element).Note that the computer should be powerful enough to run the workflows (at least 8Gb of
RAM is required). The whole package
requires about 230-240 Gb of disk space (see details here).
Trimmomatic and FastQC tools, included into the sample workflows, require Java
Runtime Environment to be installed and configured in the UGENE Application Settings. The other
tools are provided for macOS and 64-bit
Linux operating systems only.
- Transcriptomics: a new tool StringTie for transcript assembly was integrated as a workflow
element. Also, another element “StringTie
Gene Abundance Report” was implemented. It allows one to get a common report for several input
samples.A sample workflow with the
elements is available from the main menu “Tools > NGS data analysis > RNA-Seq data analysis”:
 Create a new dataset for each sample (a sample here may
be a pair of FASTQ files, for
example)!Note that StringTie and TopHat tools are available on macOS and 64-bit Linux operating systems
only. Trimmomatic and FastQC tools,
included into the sample workflow, require Java
Runtime Environment to be installed and configured in the UGENE Application Settings.
Create a new dataset for each sample (a sample here may
be a pair of FASTQ files, for
example)!Note that StringTie and TopHat tools are available on macOS and 64-bit Linux operating systems
only. Trimmomatic and FastQC tools,
included into the sample workflow, require Java
Runtime Environment to be installed and configured in the UGENE Application Settings.
However, an approach for selection in the Sequence View was also modified. Selection of an annotation was detached from selection of a sequence region. Thus, one mouse click on an annotation selects the annotation only. To select both the annotation and the corresponding sequence region double-click on the annotation.
The selection appearance was also improved:

Check the issues tracker for technical details.
- A new mode for a sequence editing appeared in the Sequence View. To switch on the editing mode click the
Edit sequence button on the left
toolbar of the Details View. Settings for handling the corresponding annotations are available
through Actions – > Edit – > Annotations
settings on sequence editing item in the main menu.

- A new mode for amino acid translations visualization in the Sequence View.

- Handy resizing of a selected region in the Sequence View and the Alignment Editor

- The default appearance of a sequence in the Details View has changed: translation frames are hidden.
- Possibility to decrease the Detail View vertical size has been added.
- Highlighting of the extended alphabet characters in the Sanger Reads Editor has been corrected.
- Other bug fixes.
See also the following documentation chapters:
- Editing Sequence
- Translating Nucleotide Sequence
- Selecting Sequence Region
- Selecting Alignment Region
Check the issues tracker for technical details.
- Tweaking of the Sanger reads mapping feature, based on feedback received.

- Fix of a critical issue of searching in amino acid translations of a nucleotide sequence.
- Other minor bug fixes and improvements.
Check the issues tracker for details.
- Support of Vector NTI/AlignX format.
- Improvement of the Sanger reads mapping algorithm.
- Bug fixes and minor improvements.
Check the issues tracker for details.
-
Sanger Reads Editor – a brand new framework for analysis of Sanger sequencing results. The main
features are:
- Reads mapping to a reference sequence.
- Browsing of the result alignment with trace chromatograms.
- Reads editing.
- Exporting of the results.
See this introductory video for the quick start.

- Bug fixes and minor improvements.
Check the issues tracker for details.
New features, improvements, bug fixes
- Fixed reported crashes with ORF auto-annotations
- Assembly Browser: better & faster coverage
- Integrated new tool: CAP3
- New plugin: DNA flexibility
- Check the issues tracker for details.
New features, improvements, bug fixes
- New plugin: Assembly Browser
- Sequence View: added functionality to manage number of visible rows in Zoom View
- Circular View: added map of restriction sites
- Phylip plugin: added tree bootstrap support
- Biostruct3D viewer: introduced structural alignment visualization
- Automatic annotation highlighting is provided for ORFs
- Check the issues tracker for details.
New features, improvements, bug fixes
- Added auto-annotation high-lighting capabilities
- Added support of export annottions in GFF format
- Added support of lauching workflows in separte processes
- Added multiple optimizations to Genome Aligner
- Fixed problem with project export
- Fixed bug with remote requests to NCBI CDD
- Workflow Designer: added support of FASTA files as queries for Smith-Waterman search
- Workflow Designer: added export/merge annotations workers
- Workflow Designer: fixed bug with iterations (append and rename)
- Added version string and estimated size to uninstall info in Windows “Add/Remove programs”
- and more minor bugfixes… Check the issues tracker for details.
New features, improvements, bug fixes
- Circular view: zooming added
- Export of rev-comple annotations fixed
- MSA dialogs: GUI fixes
- Enzymes: fixed search in range option
- Workflow Designer: fixed workers promts
- Workflow Designer: openning schema with no meta info fixed
- Worfklow Designer: added ordering to element options
- Workflow Designer: added element “Retrive data from remote DB”
- Query Designer: added strand direction parameter to queries
- Supported pDRAW document format
- Added option to export group of annotations or annotation table to CSV
- KAlign crash fixed on amino MSA
- Cloning in silico: added option to edit fragments
- Cloning in silico: added option to annotate new fragments
- Cloning in silico: fixed incorrect ligation
- and more minor bugfixes…
New features, improvements, bug fixes
- Cloning in silico: digest into fragments
- Cloning in silico: construct molecule
- Query Designer tool introduced
- External tool support: BLAST+
- External tool support: TCoffee
- OpenCL support added on Linux and Windows
- Logging system improvements
- Popup notification system added
- Fixed several “Export from annotation features”
- “Fetch by ID from Annotation” feature added
- Fixed sequence viewer behavior
- Improved circular sequence detection
- “Replace sequence” feature added
- MSAEditor: export to aminoacids
- MSAEditor: reverse-complement sequence
- Genome Aligner: save index in external file
- Workflow editor: new human-readable schema format
- Dotplot plugin: zooming options added
- Dotplot plugin: selection improvements
- Crash handler improvements
- Project view: active document is highlighted
- Fixed 3′-5′ annotations export
- Fixed BLAST annotation result reporting
- Fixed external tools managment issues
- and much more bugfixes…
New features & plugins:
- Support for external Blast: build and search
- New format is supported: MEGA
- New improved plugin system
- Support for large (over 1 GB) files analisys in DNA Assembly
Improvements:
- Crash handler is improved: now you can send us report is something unexpected happend
- Command line interface improvements: comprehensive help-system and verbose error diagnostics
- Editing script workers in workflow designer
- Deleting and hiding script workers
- Drag and drop from Project View
- Task report in Primer 3
- Application supressed exit
- Disabling of unused ports in Workflow Designer
Bugfixes:
- Fixed crash when calculating surface for large proteins
- Workflow designer fixes in GUI and usability
- Fixed crash while opening project
- Fixed crash with alignments consisting only of gaps
- Fixed crash with MUSCLE region alignment
- Fixed data formats: srfasta, sam
- Multiple designer workers fixed
- Remote tasks in Muscle and HMM are converted to Workflow schemas
- and much more…
New features & plugins:
- New plugin: cloud support. Running computational workflow on Amazon EC2;
- New plugin: external tool support. Multiple alignment with Mafft and clustal;
- New format supported: Nexus (read & write);
- Advanced scripting capabilities in Workflow Designer;
Minor improvements:
- Added capability to change name of sequence and set name when creating new sequence from text
- Added capability to cancel the insertion of workflow sample
- Items “Lock document for editing” and “Unlock document for editing” are not shown for documents wich can’t be changed
- Documents with unknown type (i.e. workflow schema) are not loaded in project
- If during loading of document error occures, document is deleted from project
- Added capability to export sequence with annotations
- Added capability to open several files
- Added capability to remove columns with certain amount of gaps(absolute or relative) in alignment editor
- When annotations are imported from CSV file, they are automatically added to active sequence view (if possible)
- When creating connection between blocks in workflow, to port by default added data from nearest block
- Automatic creation of the new project before the construction of the new Dotplot
- Added feature to search inverted repeats, search simultaneously both types of repeats
- Added feature “Lock dotplots” for simultaneous viewing several dotplots built on the same sequences
- Synchronization with PanViews of the sequences
Bugfixes:
- Annotations highlight enable/disabled fixed
- Incorrect sequence reading in Find repeats in Workflow
- Issue with saving a copy of document
- Crash when deleting three ot more documents from project
- Incorrect search in Alignment Editor
- Crash when opening incorrect file to build HMM2 profile
- Issue with creating new annotation when selected nonexistent document for storage of annotations
- Bug when call –help for nonexistent parameter in command line
- Reading alignment file with non-alignment extention for build weight matrix
- Incorrect removing of region in alignment
- Incorrect offset values in alignment editor
- Issue with reading alignment with gaps in SAM format
- Incorrect adding gaps to alignment
- Counter for annotations in annotation group
- Wrong behavior of “zoom to range”
- Toolbar disappearance after removal of the document
- Creation of cyclic schemas in workflow designer
- Fixed a bug related to incorrect positioning of the cursor
- Crash after deleting sequences from the project with opened circular view bug fixed
- Crash after selecting sequences in the locked mode with builded circular view
- Resizing circular view bug fixed
- Hidden sequence view bug fixed
- Exporting circular view as image bug fixed
- Anaglyph view option is disabled if the rendering device doesn’t support it.
New features & plugins:
- Dot plot view: effective sequence visual comparison method;
- SAM format support and PHRED quality scores import;
- Advanced chromatogram analysis. Find out details in the tutorial;
- BowTie assembly options integration, new modes for UGENE Genome Aligner. Now available from Workflow Designer!
- Weight matrix based analysis improvements;
- New Workflow Designer samples and built-in command line schemas;
- Circular view: support for annotations with virtual gap.
New features & plugins:
- Major renaming: congene -> ugene, ugene -> ugeneui
- New plugin: genome aligner;
- New plugin: bowtie;
- Dna assembly to reference functionality added;
- Poistion-weight matrix advanced support added;
- MSAEditor: new zooming mode added;
- Circular view improved.
- Overview added to Sequence View;
- Integration with JASPAR and UniProbe.
- Alignment logo added.
- Command line interface improved.
- Command line version Workflow designer support added.
- Remote request re-worked.
- Scripting framework introduced in CSV import.
- Tree viewer re-worked, new modes added.
New features & plugins:
- New plugin: Circular view;
- Export plugin: back translation feature added;
- Importing annotation tables from CSV has been implemented;
- MSA Editor: consensus mode added, usability improvements;
- Phylogenetic tree view improvements;
- Workflow designer: added support for launching tasks remotely;
- Enzymes plugin: added support for creating custom enzyme databases in BAIROCH format;
- Binary Linux distribution is available for download;
New features & plugins:
- KAlign: new plugin for multiple alignment based on popular tool;
- New format support: GFF;
- Phylogenetic tree viewer improvements: support for Newick format;
- Phylip plugin: distance matrix models added;
- Proxy authentication added.
Bug fixes:
- Smith-Waterman algorithm runs now properly from GUI;
- Fixed memory managment in Muscle plugin;
- Document deletion from Project View issue solved;
- Document format detection improved;
- MacOS version is distributed now as DMG image.
New plugins:
- Phylip plugin for building phylogenetic trees. Based on original Phylip 3.6
package.
- Neighbor Joining algorithm;
- Phylogenetic trees visualization;
- Integration with Multiple Alignment Editor.
- Distributed computing in UGENE
- Support for launching HMMER 3, Smith-Waterman, MUSCLE tasks on remote machines.
- Remote tasks support;
New features and bug fixes:
- Search for tandem repeats added introduced in Repeat Finder plugin;
- Project files transferability support;
- “Export project” feature;
- Remote databases interaction: NCBI, PDB, Swiss Prot;
- BioStruct3D plugin: anaglyph stereo mode;
- Sequence editor: several minor improvements and bugfixes;
- Lots of minor stability and usability fixes.
New plugins:
- MUSCLE 4 – a port of the newest tool for multiple sequence alignment by Robert C. Edgar;
- CUDA Support – an utility plugin which makes possible using NVIDIA graphical processing units for accelerating computational algorithms (Windows only).
New features and bug fixes:
- CUDA-accelerated Smith-Waterman algorithm is ready to use for Windows users;
- The one of the most awaited features: sequence editing has been added;
- Primer 3 integration has been finished – a complete set of preferences is available now;
- Exporting annotation tables to CSV has been provided;
- New workers have been added to Workflow Designer: Sequence splitter and Remote request;
- Collecting statistical reports feature has been added. See the dialog on the first launch of v1.5.2;
- HMM2 models support has been added to HMM3;
- Filtering by hits number has been added to enzymes search;
- Now UGENE drops ‘fantom’ documents on project closing;
- Bug with incorrect handling of sequence range selectors has been fixed in SSP dialog;
- SSP algorithms, Primer3 and PDB parser now work correctly when launched in parallel;
- A serious bug affecting Fedora users has been fixed in PSIPRED.
New plugins:
- BALL – adds new types of molecular surfaces to UGENE 3D viewer
New features and bug fixes:
- New feature: sequence line view and MSA editor now support saving fancy screenshots
- Congene crash on windows has been fixed
- HMM3 crash with malformed models has been fixed
- Crash with simultaneous primer3 tasks has been fixed
- Remote request is now stable when launched on Linux systems
- Missing Smith-Waterman search is restored
- Primer3 tasks now support cancelling
- Choosing selection color has been added to 3D viewer
- Sequence selection in Detailed View has been fixed a bit
New plugins:
- HMM3 – modern set of tools for HMM profiles build and search, based on HMMER 3.0b2
- Primer3 – integration of the primer3 tool for PCR primers design
- GOR IV and PSIPRED – protein secondary structure prediction tools
- congene – command-line version of UGENE which allows user to launch workflows from the command-line
New features and bug fixes:
- BioStruct3D viewer: molecular surface visualization
- Loading remote documents from PDB, Uniprot, Swissprot
- ‘Create document from text’ feature
- Annotations editing significantly improved
- Some critical bugs in Qt 4.5.0 are workarounded
- Stability fixes for parallel MUSCLE: deadlock prevention
Improvements and bugfixes:
- Introduced Undo/Redo support for MSA Editor
- Search in MSA Editor now works more accurate with different case of pattern
- Extra filters for SITECON search procedure added
- Now UGENE is capable of reading CLUSTAL X files
- Smith-Waterman plugin became more accurate
- Some concurrent-related bugs in MUSCLE fixed
- Some Qt 4.5 compatibility issues fixed
- Lots of minor stability fixes
Improvements and bugfixes:
- Color schemes support in Alignment editor
- A camera lock option for multiple 3D protein models
- Sequence viewer optimized to be used with large amount of annotations (up to 1 million)
- Sequence viewer can show qualifier values instead of annotation names now
- Chromatogram viewer: quality values are now shown by default for ABIF files
- Several stability and compatibility issues fixed in BLAST, MUSCLE and HMMER modules
- A number of minor stability fixes
New features:
- New plugin: Smith-Waterman. A complete implementation of Smith-Waterman algorithm
- New plugin: Restriction Enzymes. Marks restriction enzymes in a sequence. Supports ‘bairoch’ file format provided by REBASE database
- New plugin: Repeats Finder. Searches direct and invert repeats in DNA sequences. Optimized for huge sequences
- New plugin: DNA Statistics. Exportable statistical reports
- Workflow Designer: runtime state visualization of executed schema
- Workflow Designer: integrated support of workflow samples
- Workflow Designer: resizable items + snap to grid, customizable colors and fonts.
- MSA Editor: show non-gapped offsets for each sequence in alignment
- MSA Editor: a tool to search patterns in alignment
- Sequence view: support scale locks and synchronization for different sequences
- Sequence view: support for arbitrary number of rulers created by user
- BioStruct3D viewer: export of 3D PDB and MMDB models into 2D vector format (SVG)
- BioStruct3D viewer: a fast and extensible way to link active 3D model to Web resources
- Project view: supports 3 different view mode: grouping data objects by type, by document and flat view
- Project view: list of data objects for unloaded documents is shown
- Performance: UGENE now allows to specify ideal count of CPUs, threads and memory to be used
- DNA Export plugin: export of arbitrary multiple sequence alignments to FASTA format
- New file formats supported: FASTQ, MMDB (plain text ASN)
- Support for large (> 10Gb) files indexing for a fast data access
- Custom application GUI themes supported. Windows version of UGENE uses Office GUI theme by default.
Improvements & bugfixes:
- MUSCLE plugin: part of the original algorithm was optimized to utilize multicore CPUs.
- HMMER plugin: SSE2 implementation of the algorithm was added;
- HMMER plugin: hmmsearch now works in parallel mode for a single sequence by default (gives >30x boost for quad-core CPU when used with SSE2)
- SITECON plugin: >50 new prokaryotic profiles to recognize transcription factor binding sites added
- Most the views and dialogs got performance and usability improvements
- Workflow Designer: now preserves additional info of processed data (like accessions, source etc) throughout overall computation.
- Workflow Designer: write tasks now provide controls to override existing target file (either overwrite, append or rename).
- Project view: supports relocation and copying of documents.
- Annotation creation: new annotations now can be added to an unloaded annotation table object. The object is automatically loaded in this case.
- MSA Editor: allows to change fonts, initial and limited support for zooming operations
- BioStruct3D viewer: support for single structure with multiple 3d models
Improvements and bugfixes:
- A QT4.4 bug that prevents correct rendering of OpenGL widgets on some Linux platforms is work-arounded.
- Group naming erorrs when nested groups used is fixed.
- A new and more complete implementations for ABIF and SCF formats added.
- Fixed bug that prevented ‘z’ char to be used in annotation names.
- Annotations tree widget performance improved.
- Sitecon plugin now processes custom regions correctly.
- Workflow Designer crash when multiple iterations are used is fixed
- For very short DNA sequences without ‘U’ and ‘T’ letters DNA genetic code is used now by default instead of RNA.
- ‘Tab’ characters in FASTA and Genbank plain text formats do not produce error messages anymore.
- Ubuntu package of UGENE was split into 2 packages (data and gui) to satisfy Ubuntu packaging rules.
Improvements and bugfixes:
- Fixed issue that can disallow to open more that 1 view on some 64-bit systems.
- Fixed crashes when several PDB models are opened side by side in a single 3D-viewer window.
- Fixed crash when opening ABI file with a very short sample name.
- Added “Check For Updates” menu.
- Fixed crash in “Find pattern” dialog when complement strand is selected for amino sequences.
- Fixed chromatogram objects association for ABIF and SCF files.
- Fixed genetic code autodetection for PDB file format.
- Improved docked labels appearance for Linux and Mac OS X platforms.
- Improved ‘Add object to view’ menu logic.
New features:
- Linux support. Packages for Fedora and Ubuntu
- New plugin! Workflow Designer: allows to design custom computational workflows
- New plugin! SITECON: search for Transcription Factor Binding Sites (TFBS) in DNA sequences
- New plugin! BioStruct3D: 3D molecular protein and DNA structure viewer for PDB format
- Stockholm and PDB formats support
- Browsing multiple sequences side by side in a single sequence view window
- Sorting by annotation qualifier values
- ‘db_xref’ qualifiers made interactive and are linked to corresponding web databases
- Translation and complement strands can be hidden in sequence view
- Support for localization. English and Russian translations are included into the default distribution
- Tasks can generate detailed reports now
Improvements & bugfixes:
- Task progress is now shown in status bar
- Critical fix: Memory leaks and crashes fixed in code that integrates MUSCLE package into UGENE
- Critical fix: Default parameters of HMMER-related dialogs are now the same as in original HMMER
- Logs view now do not show traces by default
- Drag&drop support for all file types supported by UGENE
- >100 other minor fixes, usablity and performance improvements !
New features:
- MacOS v10.4 and v10.5 support.
- Multiple sequence alignment editor. MUSCLE alignment algorithm integrated as a separate plugin.
- New plugin! ORF Marker: Searches for ORFs in DNA-sequences.
- New plugin! ChromaView: The plugin integrates chromatogram viewer into ‘Annotated DNA View’
- New plugin! Remote request: Annotates selected DNA-sequence regions by sending requests to BLASTn, BLASTp and CDD servers. Can be extended to use other servers with scripts.
- New plugin! UMUSCLE: Adds MUSCLE alignment algorithm to the system.
- Files located on remote servers can be added to project by HTTP protocol
- Scripting support for custom WEB-based BLAST/CDD-like queries. Binding for all GUI/Utility classes and ability to create any algorithms using script.
- DNAGraphPack plugin supports 4 new graph types: GC & AT deviations, Karlin Signature Difference and Informational Entropy
- All genetic translation tables of Genbank database are supported and can be selected by user.
- SCF format support added.
- Support for unloading documents without removal them from a project.
Improvements & bugfixes:
- UGENE can be associated to open all file types it supports from command line. File binding comes by default with Windows installer.
- Better integration of uHMMER plugin with Annotated DNA View and MSA Editor
- DNAGraphPack plugin now shows interval based graphs for large zooms to keep min/max information precise.
- Drag&Drop functionality between ‘Project View’ and ‘Annotated DNA View’
- Dozens of GUI, performace and compatibility improvements and bug fixes.
New features:
- EMBL, ABI, CLUSTALW and raw sequence file formats supported.
- Custom document format settings option added when adding documents to project. Use this option if you need to merge all DNA sequences found in document into a single sequence.
- New plugin: uHMMER . This is a port for hmmbuild, hmmcalibrate and hmmsearch tools from HMMER2.3.2 package
- New plugin: DNA Export. This plugin adds functionality to export various types of DNA sequence selection to FASTA file format.
- Starting from this version UGENE works transparently with compressed documents in GZIP format. For example, compressed files can be added to project.
- New plugin: DNA Annotator. This plugin adds annotation based searches to runtime. Current version allows to find regions that contain a predefined set of annotations.
Improvements & bugfixes:
- Multi-selection support added for annotations tree used in ‘Annotated DNA View’.
- Annotations’ colors, placement and visibility properties were made customizable.
- “Find DNA sequence” algorithm was tuned. Several bugs fixed.
- A lot of minor GUI and performance improvements.