
UGENE 1.15 List of Features
This article is a brief overview of the UGENE 1.15 new features.
We have improved the UGENE’s ability of working with shared databases. It is possible now to use the databases in your workflows. Workflows can use databases in both ways: read data from a database and write data to it.
PCR
The new UGENE version provides the In silico PCR feature. You can design primer pairs and simulate your PCR experiment before going to a wet lab. This is an easy way to safe your time using UGENE as a primer design tool.
You can keep a library of your primers. Add primers into the library and they will be stored there between UGENE sessions. The primers from the library could be used for running In silico PCR. You can actually build a library of primers for you lab and share it between UGENEs or other primer design tools. Whenever a new sequence you want to amplify comes in you might check it with you pre-built primer library.
Search
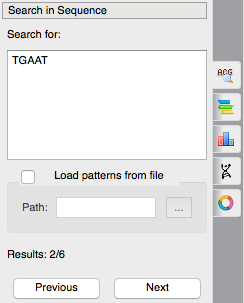
The feature of subsequences searching has become more convenient. Now you can search a subsequence like you do it in web-browsers by ctrl+F hotkey. You just input your subsequence and immediately see the result of your search, you can navigate through the results with Prev-Next buttons. Enter a target subsequence, navigate through the results and then create annotations if you want.
Most of sequence algorithms in UGENE have been improved for working with circular sequences. The algorithms are Smith-Waterman searching, local and remote BLAST, ORFs searching, Primer3 and others. If a sequence is circular then the algorithms automatically consider this property during the calculations.
We have added the new options panel for adjusting the circular view of sequences. Now you can change many options for getting the needed picture for you publications. You can edit colors, fonts, sizes of the annotations, plasmids and select a way how tooltips are shown.
de novo Assembly
The long-awaited feature, de novo assembly tool, has finally appeared in UGENE. We integrated the SPAdes de novo assembly instrument. This assembler is good in assembling of single cell and multi-cells bacterial data sets. The SPAdes assembler is available in the special genome-assembling dialog and in the Workflow Designer. In UGENE we simplified the way the input reads are specified. You will find a dialog that helps you figure out how to input libraries and pairs of reads. Pay attention that SPAdes genome assembler requires a lot of RAM for big genomes.
It is possible now to export the NGS coverage of assemblies to a file. The exported text file contains a table that describes NGS coverage – the count of aligned short-reads for each assembly position.