
Tutorial: Alignment of Short Reads with UGENE
In this block we will perform short reads alignment using UGENE. One of the applications of alignment is the gene mapping. We will do genomic mapping of a simulated dataset generated from E.coli reference genome (NC_010473). The dataset consists of 100,000 paired-end reads of size 50 bp.
Starting the Mapping
UGENE provides GUI for popular alignment tools Bowtie, Bowtie2 and bwa aln (or bwa mem). Additionally a unique tool called UGENE Genome Aligner is available for users. The aligners are accessible from the main menu item Tools->Align to reference.
In the tutorial we will learn how to align single-end reads with Bowtie from UGENE, however the interface for the aligner is general and other tools can be used in a similar manner. For instance bwa aln or bwa mem can be used here.
Bowtie is an index-based aligner, thus one has to first build index of the reference sequence before starting the alignment. One can create the index for genomic mapping from UGENE using main menu item Tools->Align to reference->Build index.
In the appeared dialog we first choose for which program we are building index (Bowtie aligner) Then it is required to set path to the reference genome sequence in FASTA format (ecoli.fa in our case). The path to the index file will be set automatically. Click OK to create the index.
In order to start the alignment activate main menu item Tools->Align to reference->Align short reads. Next select Bowtie aligner. The index for the gene mapping was already built in the previous step therefore we will select option Prebuilt index in the dialog parameters. The index file can be selected using browse button (…) next to Reference sequence field. Note, that one of the index files has to be selected, for example ecoli.1.ebwt. Next we set the library option to Paired-end and add short reads to the input data list. The correct mate order (upstream or downstream) has to be set using Order option after the reads are added. To start the alignment press Start button.
After the alignment is finished, you will be asked to import the alignment into the UGENE Assembly Viewer.
Visualizing BAM files
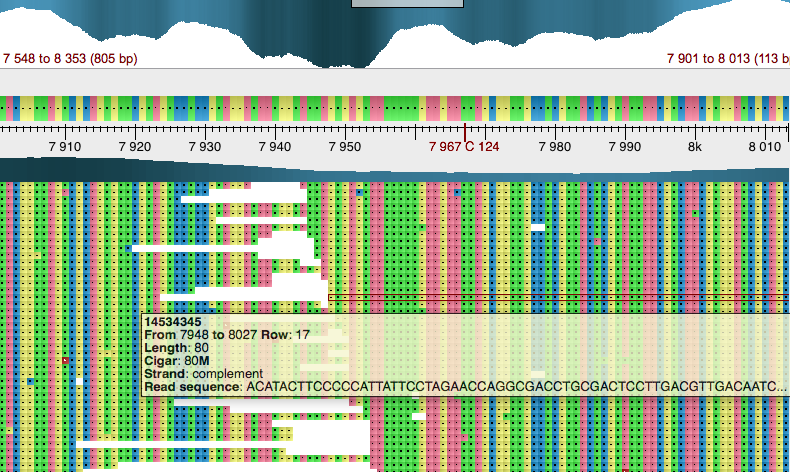
The UGENE Assembly Browser allows to view gene mapping region by zooming into them.
The following features are available:
- Navigation and zooming
- Read coloring
- Extracting alignment groups
It is possible to color reads using different coloring schemes. The default one is Difference. Other possible are Nucleotide, Alignment strand and Paired reads.It is possible to extract a group of reads or consensus to FASTA format using context menu item Export.
A screenshot of an alignment can be taken using the Export image button.
Creating pipeline for Next Generation Sequencing data using Workflow Designer
UGENE Workflow Designer includes a number of useful elements to work with Next Generation Sequencing data.
We will go through an example of creating a small pipeline for calling SNPs for the alignment files.
The SNP calling will be performed using SAMtools.
First we will open the Workflow Designer using menu item Tools->Workflow Designer
Next we will add the following elements to the Workflow:
Read Assembly and Read Sequence from Data Readers
Write Variations from Data Writers
Call variants from NGS: Variant Calling
Read Assembly and Read Sequence elements should be connected with corresponding input ports of Call Variants block, while Write Variation element should be connected with its output port. When required the input data ports must be edited.
We will use a previously created alignment ecoli.sam as input for Read Assembly and ecoli.fa as reference for Read Sequence elements. Additionally we will set output path for the Write Variations element.
Next we will click Run to start the Schema.
The resulting SNPs can visualized in the Assembly Editor by adding a SNP track.
RNA-seq analysis with Tuxedo pipeline
In this block we will perform gene expression and transcript reconstruction analysis from RNA-seq data.
UGENE includes widely-used RNA-seq analysis pipeline Tuxedo. The description of the pipeline can be found in the publication.
We will use the example data from this publication available from the GEO-accession GSE32038 . The genome and annotations can be downloaded from iGenome collection.
To activate the pipeline go to Samples->NGS and select RNA-seq analysis with Tuxedo.
The interactive wizard will guide the setup of the pipeline. We will select "Normal pipeline" and "Paired-end reads".
Next we will set the path to the analysed sequencing reads. In this demonstration we will use only one sample for first and second mates respectively: GSM794483_C1_R1_1.fq.gz and GSM794483_C1_R1_2.fq.gz.
We will use Bowtie1 in Tophat settings. The BowtieIndex can be found from the iGenome archive.
For Cufflinks we will provide the path to Reference annotation in GTF format.
After that we click run to start the workflow. The results workflow can be loaded and view in UGENE.