
UGENE Mini-Tutorials: Open Databases, Genome Mapping and More
Separate a set of primer sequences
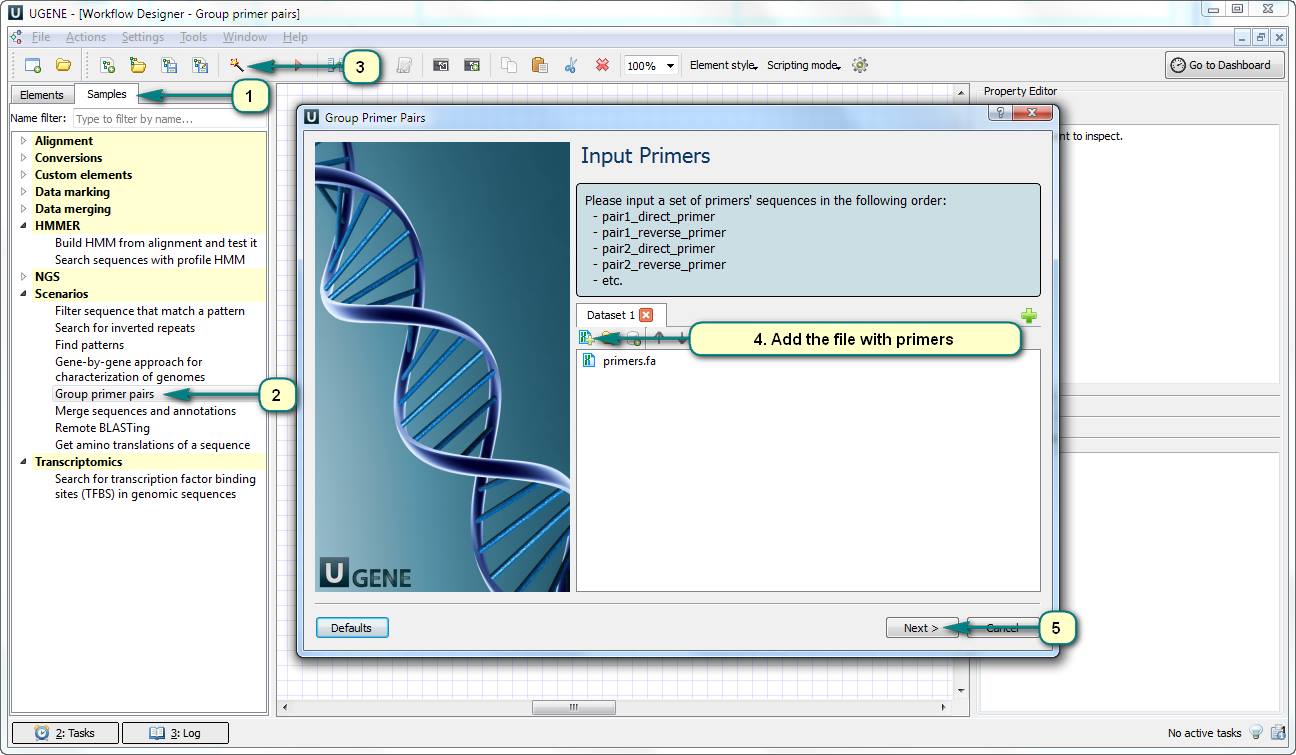
How to separate a set of primer sequences into compatible groups?
If you want to do a set of PCR reactions for amplifying of several sequences (e.g. for extracting of a set of genes), you can save your time and money doing several PCR reactions in one experiment. However, some pairs of primers could be incompatible forming dimers. UGENE Workflow Designer can help you with separating the primers into compatible groups.
Open Workflow Designer and use the predefined sample “Group primer pairs”. The input data of this workflow is a file with primer pairs (e.g. in the Multi-FASTA format): direct primer of pair 1, reverse primer of pair 1, direct primer of pair 2…
The result of the workflow is the report file – a table with the groups of primer sequences. Each couple of primers in a group does not form a dimer.
Download data from open databases
How to download data from open databases via the command-line interface?
The UGENE command-line interface provides a set of ready-to-use solutions for different bioinformatics tasks. One of the tasks is getting data from the open databases like NCBI GenBank, PDB, Swiss-Prot, etc.
If you want to download data, run the following command:
ugene -task=fetch-sequence -db=database-id=<;-separated list of identifiers>
The available databases are NCBI GenBank (DNA), NCBI GenBank (Protein), PDB, Swiss-Prot, UniProt.
For instance, run this command for downloading two nucleic sequences NC_001363 and D11266 from the NCBI GenBank database:
ugene –task=fetch-sequence –db=genbank –id=NC_001363;D11266
After that the data will be downloaded and saved in the current directory. For saving the data in a directory you want, use the parameter -save-dir=path.
Here you can find the documentation about this solution.
Genome mapping
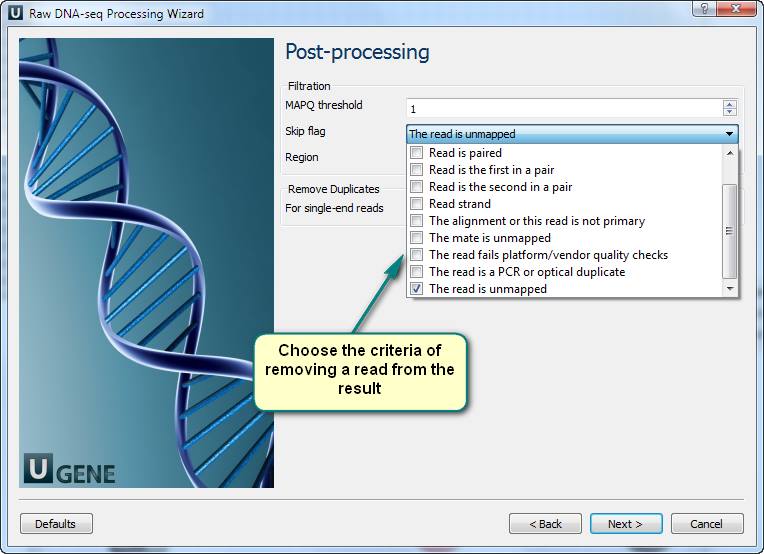
How to get a good genome mapping (alignment) from raw NGS data?
At first glance, elementary analysis of sequencing data (short-reads genome mapping) is simple. But if you just map short-reads to a reference sequence using one of the mapping tools (e.g. Bowtie or BWA), it wouldn't be enough. If you want to get an alignment of good quality, you have to preprocess the short-reads, filtering them and trimming their low-quality ends, and filter the mapped and unmapped short-reads.
UGENE Workflow Designer provides a ready-to-use NGS pipeline for managing the whole cycle of raw Illumina sequencing data processing. This workflow includes:
1. Filtering of short-reads by the CASAVA header.
2. Trimming of the low-quality ends of the short-reads.
3. Mapping of the short-reads to a reference sequence using BWA-MEM.
4. Filtering of the mapped short-reads using SAMtools.
For running the workflow:
1. Open Workflow Designer and double click the “Raw DNA-seq processing” sample in the “NGS” category of the “Samples” tab.
2. If your data are paired short-reads, choose the “Paired tags” item. Otherwise, choose “Single tags”. Then click the “Setup” button.
3. Now you see the workflow wizard. Following the wizard’s pages, you can set up the workflow parameters: input files with short-reads and a reference sequence, filtration thresholds, results filtration ways, etc.
Finally, you will get the NGS pipeline that is ready for analysis of your data. The result of the workflow is a set of files produced in each step of the process. The last file is the target good mapping.
Here you can find the corresponding documentation about this workflow.