
UGENE Mini-Tutorials: Workflow Designer Samples and Inverted Repeats and More
Workflow Designer samples
UGENE Workflow Designer provides the set of ready-to-use samples. Each sample solves a particular bioinformatics task. The list of the samples is available in the “Samples” tab of the Workflow Designer window.
A mouse click on a sample shows the information block about the sample, its functions, used data, etc.
A double click opens a sample. Each sample is equipped with a wizard. Using a wizard, you can easily set up the input data and other parameters of your workflow. Click the magic wand button in the toolbar for running the wizard.
You can find the documentation about UGENE Workflow Designer samples here.
Search for inverted repeats in a nucleic sequence
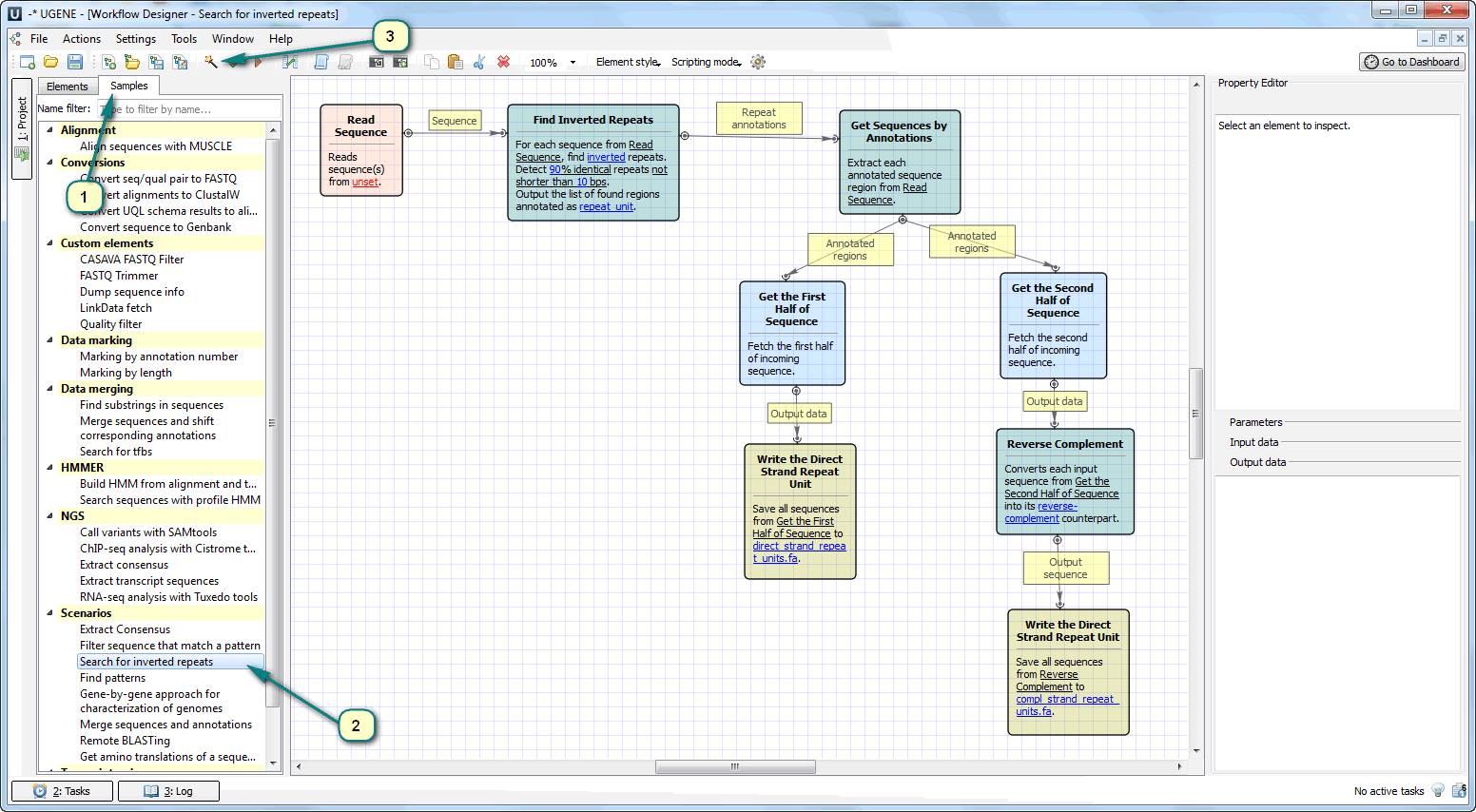
Inverted repeats are the type of single tandem repeats located in different strands: one part of a repeat is in the forward strand and the other part is in the reverse-complement strand (inverse sequence). There is the ready-to-use sample in UGENE Workflow Designer for searching such kind of single tandem repeats.
Open Workflow Designer and double click the “Search for inverted repeats” sample in the “Scenarios” chapter. Click the magic wand button in the toolbar for opening the wizards. The wizard would help you with:
1) Supplying the file with an input nucleic sequence (the inverse sequence is generated automatically).
2) Setting up the repeats search parameters: minimal repeat length, repeats identity, repeats distance, etc.
The result of this workflow is two files with the found repeats sequences: a file with the forward parts of repeats and a file with reverse-complement ones.
How to convert a multiple sequence alignments file into a file of a needed format
If you use the command line interface, you can run different tasks in UGENE using this interface. For example, you can convert multiple alignment files into files of needed formats. The command you are about to run turns UGENE into a fasta format converter.
Run UGENE with the following parameters:
ugene –task=convert-msa –in=input_file_path –out=result_file_path –format=format_name
Here is the list of possible values of the “format_name” parameter: clustal (default value), stockholm, msf, mega, phylip-sequential, phylip-interleaved, and fasta (to create a fasta format converter).
The following command shows the help message about sequence alignments conversion:
ugene –help=convert-msa
The corresponding documentation page.
The list of tasks that could be run via the command line interface.