
История изменений
- В UGENE появился темный режим.
- Диалог поиска сайтов рестрикций притерпел ряд визуальных улучшений.
- Поле фильтрации в диалоге поиска сайтов рестрикций теперь поддерживает множественные запросы, разделенные запятой.
- Увеличен уровень общей стабильности приложения — исправлено более 20 ошибок.
- Добавлена возможность делиться файлами, хранящимися на платформе workspace.ugene.net, прямо из UGENE.
- Реализована поддержка инструмента Kraken для анализа данных.
- Обновлена цветовая схема редактора множественных выравниваний.
- Исправлены проблемы совместимости для окружения Linux с предустановленным Qt.
- Улучшена поддержка high DPI на Linux и Windows.
- UGENE Cloud Workspace: workspace.ugene.net
- Улучшена поддержка фильтрации сайтов рестрикций
- Исправлены проблемы с внешним инструментом `vcf-consensus`
- Пакет Samtools обновлен до последней версии
- Добавлена поддержка сайтов рестрикции с несколькими точками разреза
- Исправлена ошибка в Phylip bootstrap, которая могла приводить к некорректным нулевым расстояниям
- Устранены несколько сбоев в Workflow Designer, связанных с точками останова
- Улучшена обработка больших наборов данных, превышающих доступную память
- Primer3 может быть запущен без целевой последовательности.
- В UGENE был реализован Primer-BLAST.
- В UGENE добавлен инструмент визуализации шпилек mfold.
- Обновлена до последней версии база аннотаций плазмид PlasMapper.
- Python, включаемый в состав UGENE, обновлен до версии 3.
- Расширен набор пресетов для Primer3.
- Улучшена совместимость с настройками темы в macOS.
- Исправлена серия проблем связанных со стабильностью работы и ошибками в интерфейсе UGENE.
Так же обратите внимание на новое видео о недавних улучшениях в UGENE.
Сэнгер файлы (.ugenedb файлы с Сэнгер прочтениями внутри), порождённые UGENE версии 50, не будут открываться в предыдущих версиях UGENE. Но при открытии "старых" Сэнгер файлов в новом UGENE версии 50 проблем не возникнет.
- Primer3 получил функцию "Пресеты", позволяющую выбирать набор предустановленных настроек для запуска инструмента. На данный момент доступно два пресета - настройки по-умолчанию и Рекомбинатозная Полимеразная Амплификация.
- Primer3 получил функцию фильтрации полученных в результате пар праймеров в зависимости от их гомо/гетерокомплементарности.
- Исправлено выделение аннотаций в Редакторе последовательностей.
- Различные небольшие исправления и улучшения интерфейса.
- Улучшена поддержка macOS: исправлены десятки мелких проблем в интерфейсе.
- Переработан интерфейс работы с сайтами рестрикций: добавлены новые фильтры и детализация сайтов.
- Обновлены до последней версии большинство внешних инструментов поставляемых с UGENE.
- Расширена поддержка форматов файлов PDB и Clustal.
- Исправлено много небольших проблем связанных с удобством и стабильностью работы UGENE на всех платформах.
- Улучшена работа с закладками. Появилась возможность обновлять закладки.
- Улучшена поддержка больших SAM и BAM файлов.
- Для расчёта температуры плавления по умолчанию используется алгоритм из инструмента Primer3.
- Улучшено описание параметров и работа инструмента Primer3.
- Исправлены проблемы с фокусом в ряде диалогов на macOS.
- База сайтов рестрикций Rebase обновлена до последней версии.
- Для сайтов рестрикций добавлена возможность выбора коммерческих производителей.
- Исправлены баги при работе с аннотациями и в многострочном режиме редактора выравниваний.
- Добавлен новый инструмент построения филогенетических деревьев: FastTree.
- Добавлен новый метод расчёта температуры плавления на основе инструмента Primer 3.
- Улучшено отображение филогенетических деревьев: зумирование, скролл, сохранение выделенного и коллапс состояний при переключении формы дерева и многое другое.
- Исправлен алгоритм создания списка регионов с хорошим покрытием в Assembly Browser для больших последовательностей.
- В редакторе выравниваний добавлена возможность экспорта изображения выравнивания в режиме переноса строк.
- Улучшена работа с определением алфавита для последовательностей содержащих символы из расширенного ДНК/РНК алфавита.
- Новый режим отображения множественного выравнивания: Wrap Mode (перенос по строкам).
- Улучшения в отображении филогенетических деревьев: возможность скрывать узлы, копирование изображения, настройка кривизны ветвей.
- Primer3 обновлён до последней версии.
- Экспериментальный функционал: добавлен новый инструмент Genecut.
- Множество других исправлений и улучшений.
- Обновление Qt с версии 5.12.12 до 5.15.6.
- Из UGENE убрана поддержка работы с CUDA и OpenCL.
- Исправлены проблемы с включением режима визуализации 'Кладограмма' и 'Филограмма' для деревьев в круговом и неукорененном режимах.
- Улучшена поддержка Unicode в именах BAM и SAM файлов, используемых модулем Samtools.
- Библиотека GL2PS для экспорта 3D-визуализаций в векторные 2D форматы была обновлена до последней версии.
- Улучшена производительность и стабильность работы парсеров для GFF и ASN форматов.
- Алгоритм построения деревьев MrBayes теперь использует идентичные параметры по умолчанию для различных запусков и производит воспроизводимые результаты.
- Множество других исправлений и улучшений.
- При открытии результатов ассемблирования UGENE автоматически выбирает и показывет контиг с ридами.
- Улучшена поддержка выравниваний с дублирующимися именами последовательностей: исправлены все доступные в UGENE инструменты для построения деревьев.
- Инструмент MrBayes при построении деревьев теперь корректно получает параметр для настройки генератора случайных чисел и производит повторяемый результат.
- В Редакторе Выравниваний улучшена работа инструмента "Приблизить выбранный регион".
- Улучшена точность выделения совпадений на диаграмме Dotplot при помощи мыши.
- Из UGENE убрана экспериментальная поддержка работы с удалённой базой данных MySQL.
- Множество других небольших улучшений в графическом интерфейсе и стабильности работы.
- Новый функционал в Редакторе Выравниваний: Список Исключенных Последовательностей (Exclude List).
- Добавлен выбор алгоритма выравнивания в меню 'Перевыровнять выделенные последовательности".
- Виджет панорамы в Редакторе Выравниваний был оптимизирован и теперь включен по умолчанию для выравниваний любых размеров.
- Улучшена скорость навигации в Редакторе Выравниваний при работе с большими выравниваниями.
- В диалоге IQ-TREE для наиболее популярных опций добавлены отдельные поля ввода.
- Двойной клик по имени последовательности в Редакторе Выравниваний перемещает между началом и концом последовательности.
- Исправлен порядок записи регионов при экспорте последовательностей для join+complement аннотаций.
- При создании новых аннотаций по-умолчанию используются имена из стандарта Genbank.
- Ускорен алгоритм маппинга ридов на референсную последовательность в случае большого числа ридов.
- Исправлена регрессия с невозможностью присоединиться к заново созданной базе MySQL.
- Улучшена поддержка формата филогенетических деревьев Newick.
- Множество других небольших исправлений и улучшений.
- Добавлен новый инструмент для построения филогенетических деревьев: IQ-TREE.
- Открытие очень больших выравниваний больше не блокирует UGENE.
- В редакторе последовательностей исправлено некорректное изменение выделения при дабл-клике.
- Добавлена поддержка Unicode для формата Stockholm.
- Улучшена работа формата FASTA для файлов с большим числом коротких последовательностей.
- ABIF файлы с некорректными датами теперь не приводят к падениям UGENE.
- Поиск большого списка паттернов в последовательности больше не заблокирован.
- Исправлен ряд багов связанных со стабильностью работы и потреблением памяти в алгоритмах поиска праймеров и Smith-Waterman.
Редактор выравниваний
- Добавлена поддержка раздельного выделения последовательностей с CTRL.
- Поддержаны все варианты алгоритмов инструмента PhyML.
- Реализована возможность перемещения последовательностей в другой документ.
- Работа с группами: показан размер и добавлена сортировка по размеру групп.
Редактор выравниваний по Сэнгеру
- Добавлена возможность просматривать альтернативные мутации.
- Исправлены ошибки вертикального скроллинга.
Редактор последовательностей
- Улучшена скорость работы и точность графиков.
- Улучшена работа режима авто-аннотирования.
- Исправлены ошибки при работе с файлами более 2Гб.
Разное
- Улучшена реализация алгоритма Smith-Waterman для CUDA.
- Добавлена поддержка Unicode для Newick, Plain text, Diff, VCF, SNP форматов.
- Утилита генерации случайной последовательности теперь работает в разы быстрее.
Новая версия содержит следующие улучшения:
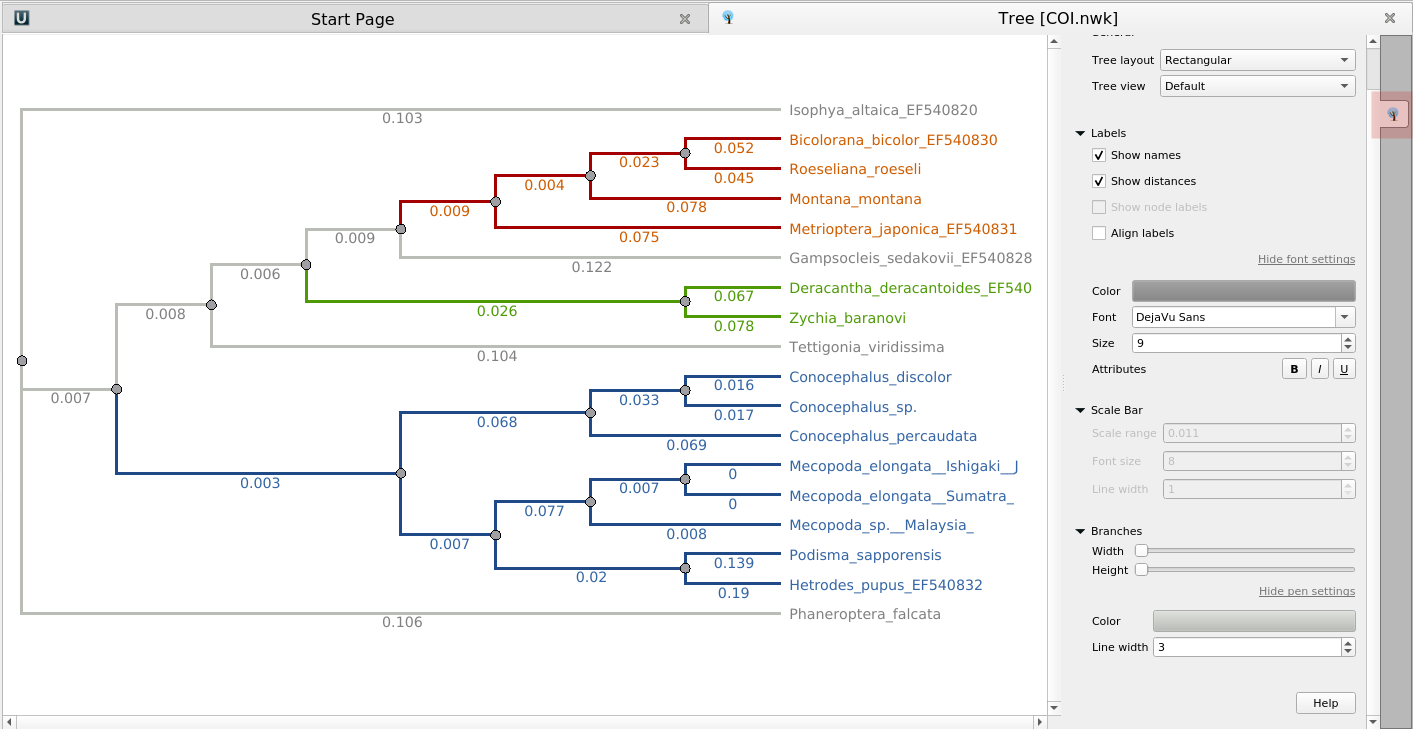
- Филогенетические деревья: независимая настройка цвета и шрифта для различных частей дерева:
- Опция "Добавить последовательность в выравнивание" получила поддержку выбора алгоритма: ClustalO,
MAFFT, Muscle или UGENE:

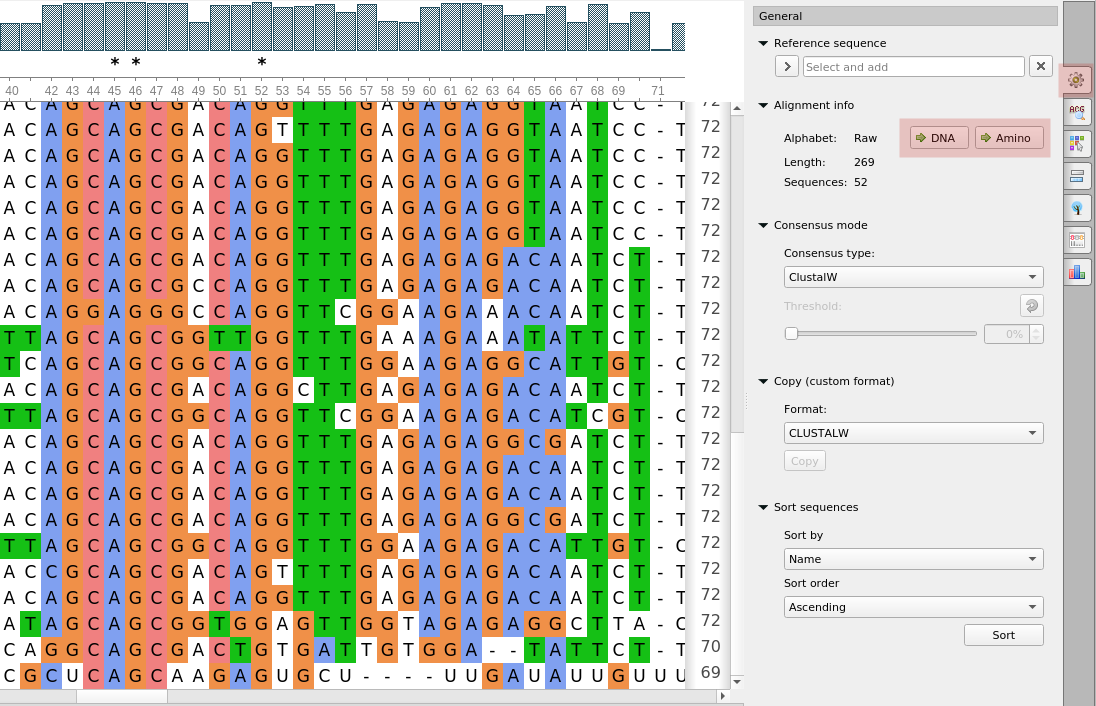
- Выравнивания с RAW (неопределённым) алфавитом могут быть "исправлены": сконвертированы в DNA, RNA или
Amino алфавит:
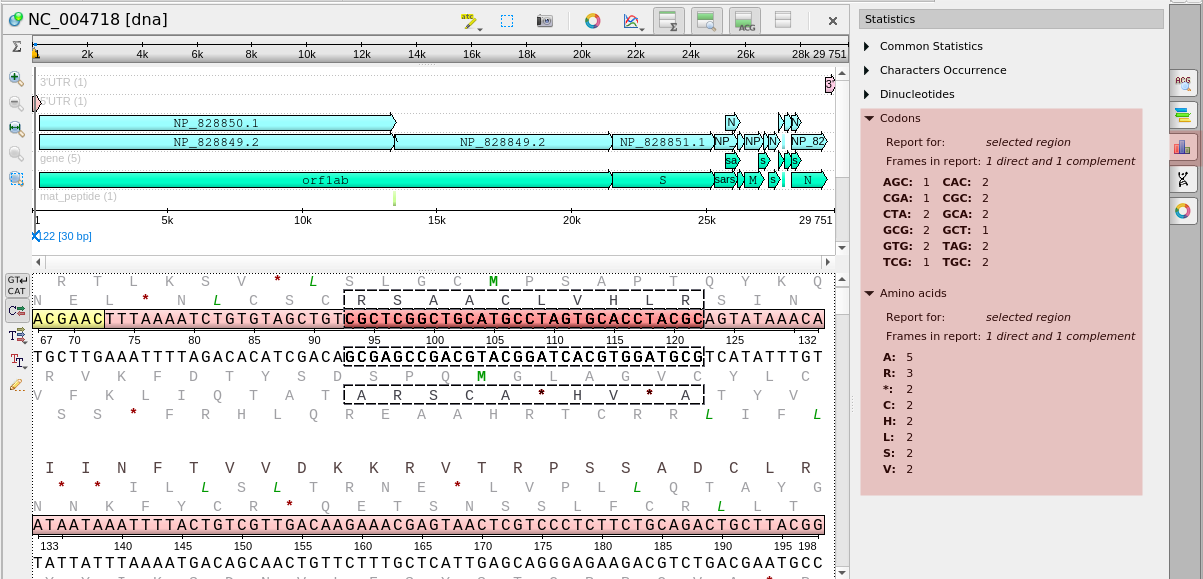
- Редактор последовательностей получил новую опцию: показ статистики кодонов и аминокислот в выделенном
участке:
- Другие изменения:
- Улучшена поддержка Unicode для FASTA, MSF и CLUSTALW форматов.
- UGENE теперь говорит и на турецком. Используйте меню Настройки/Установки для того чтобы сменить язык.
Новая версия содержит следующие улучшения:
- Экспорт аминокислотного представления:
Диалог «Экспорт выравнивания в амино трансляции» был улучшен. Теперь вы можете выбрать, хотите ли вы экспортировать гэпы или нет и если хотите, то в качестве какого символа — «X» или «-». Также появилась возможность выбрать рамку трансляции.
- Функции сортировки:
Теперь доступна функция «Сортировка по числу ведущих гэпов». Помимо этого, все функции сортировки теперь работают не только для всего выравнивания, но и для выделенных последовательностей.

- Функция «Вырезать»:
Появилась возможность вырезать выделенную часть выравнивания. Вы можете найти соответствующий пункт контекстного меню или воспользоваться сочетанием горячих клавиш «Ctrl+X».

- Другие изменения:
- Уменьшен размер минимального шрифта в Редакторе Множественных Выравниваний.
- Двойной щелчок на прочтении перемещает экран на значащие символы в Редакторе Выравнивания по Сэнгеру.
- Исправления различных ошибок и прочие улучшения.
С полным списком изменений можно ознакомиться здесь
Новая версия содержит следующие улучшения:
- Алфавитный порядок Внешних Инструментов:
Обратите внимание на то, что на вкладке «Внешние инструменты» в «Настройках» список стал упорядоченным по-алфавиту.
- Улучшен механизм копирования/вставки и именования в редакторе выравниваний:
Копирование и вставка последовательностей теперь сохраняет имя копируемой последовательности, а так же правит его для соблюдения уникальности имени. Помимо этого, появилось возможнасть вставлять последовательность в конкретное место в выравнивании (рядом с текущим выделением).

- Улучшен механизм именования в окне Закладок:
Написание имен файлов и принадлежащих им объектов сделано удобнее в окне «Закладки»

- Другие изменения:
- Улучшен механизм валидации Внешних Инструменов, что ускоряет запуск программы.
- Обновлены Внешние Инструменты Trimmomatic, BWA и Bowtie2.
- Поправлено большое количество ошибок, связанных с работой с базой данных, внешними инструментами и другими сценариями.
С полным списком изменений можно ознакомиться здесь.
В новой версии были добавлены следующие нововведения:
- Добавление ярлыка запуска UGENE на рабочий стол:
Обратите внимание на появившуюся кнопку «Добавить ярлык на рабочий стол» в меню «Помощь».
- Улучшено копирование аннотаций в редакторе выравниваний:
Копирование последовательностей выделенных аннотированных теперь привязано к направлению последовательности, на которой данная аннотация расположена. Если выделено несколько аннотаций, расположенных как на прямой, так и на комплементарной последовательности, то при нажатии кнопки копирования, будут скопированы последовательности с соответсвующих направлений.

- Другие изменения:
- Поддержка MySQL версии 8.
- Улучшена визиализация филогенетических деревьев.
С полным списком изменений можно ознакомиться здесь.
В версии 35 появились следующие функции в редакторе выравниваний:
-
Сортировка последовательностей по имени и по длине: для этого воспользуйтесь меню «Сортировка»
из
главного или контекстного меню.Также вы можете сделать это при помощи главной вкладки на панели опций:

Возможны 4 вида сортировки:
- По имени, по возрастанию
- По имени, по убыванию
- По длине, по возрастанию
- По длине, по убыванию
-
Поиск по именам последовательностей:
Для поиска по именам последовательностей воспользуйтесь вкладкой «Поиск в выравнивании» на панели опций и
выберите соответствующий контекст поиска.

-
Группировка последовательностей: Для поиска паттерна в выранивании или в именах
последовательностей
воспользуйтесь вкладкой «Поиск в выравнивании» на панели опций.Чтобы сгруппировать найденные результаты нажмите кнопку «Группировка». Результаты будут выделены и
и сгруппированы вверху после первого нажатия на кнопку «Группировка» или внизу после повторного нажатия
на нее.
-
Новая цветовая схема «Слабая гомология»: Когда есть выбор между двумя символами и не
указаны другие
правила по окраске используются следующий приоритет: T>U>G>C>A>B>D>H>K>M>R>S>V>W>Y>N.Правила окраски:
- Не окрашивать гэпы («-«), т. е. используется чёрный шрифт и белый фон.
- Остатки должны быть окрашены в соответствии с их процентным числом в столбце.
- Остатки с наибольшей частотой/приоритетом в столбце.

-
Другие изменения:
- ClustalO обновлён до последней версии.
- Меню «Навигация» и «Внешний вид» в редакторе выравниваний стали более удобными.
- Меню «Редактировать» для последовательностей стало иметь более удобную структуру.
В версии 34 появилось множество новых функций в редакторе выравниваний:
-
Возможность перевыравнивания последовательности на остальные последовательности в выравнивании:
выделите в выравнивании одну или
несколько последовательностей и нажмите
 to other sequence' button on the AE toolbar") на панели инструментов.
Выделенные последовательности будут выровнены на оставшиеся.
на панели инструментов.
Выделенные последовательности будут выровнены на оставшиеся.
-
Расширенный набор опций для поиска: в новую версию добавлены новые
возможности поиска в выравнивании: поиск одновременно нескольких паттернов, использование других
алгоритмов помимо «Exact» (т. е.
точного совпадения), возможность искать только в заданных колонках. Новые опции доступны на вкладке
панели опций аналогично редактору
последовательностей.

- Улучшенный внешний вид выделенного участка выравнивания: выделенный участок более яркий по сравнению с остальным выравниванием, более чётко выделена граница участка.
- Выделение того же участка в другой последовательности: если в открытом выравнивании выделен некоторый участок и требуется выделить аналогичную область в другой последовательности, используйте горячую клавишу Ctrl (или Cmd на macOS) и нажмите на имя нужной последовательности.
- Исправление ошибок при работе с «collapsing mode»: починено несколько ошибок при работе с выравниваниями со «схлопнутыми» одинаковыми последовательностями.
Изменения в редакторе последовательностей:
- Сохранение конфигурации внешнего вида: теперь изменения внешнего вида редактора будут сохраняться при открытии новых окон, даже после перезапуска программы. Например, сохраняется состояние Zoom View (показывать/не показывать) или отображение всех рамок считывания.
-
Статистика по последовательности: исправлен подсчёт статистики для нуклеотидной
последовательности или её выделенного участка, добавлена
информация по двухцепочечным молекулам, см. документацию
(на английском языке).

- Копирование/вставка аннотированных участков: по запросу от пользователя для удобного аннотирования последовательности добавлено несколько опций копирования аннотированных участков (по аналогии с просто выделенными участками последовательности).
- Исправление ошибок, возникающих при работе с круговой последовательностью: исправлено несколько ошибок.
Прочие изменения:
- Поддержана новая версия формата SWISS-PROT.
- Оставлены только BLAST+ и NCBI BLAST, устаревшая версия BLAST более не поддерживается.
- Уменьшилось количество поддерживаемых пакетов, пакеты для 32-битных операционных систем более не поддерживаются. Старые пакеты доступны тут.
- Пакеты для операционной системы Windows подписаны цифровой подписью Унипро.
Новая версия содержит различные улучшения по добавлению новых инструментов в UGENE:
- Импорт вашего инструмента в интегрированные внешние инструменты UGENE: список внешних инструментов доступен в настройках приложения. Для импорта вашего инструмента см. документацию.
- Обновлен диалог-помощник для конфигурации запуска инструмента: для использования инструмента в
дизайнере вычислительных схем UGENE
создайте соответствующий элемент вычислительной схемы, задающий входные/выходные данные, параметры,
команду запуска инструмента. Чтобы открыть
диалог-помощник, нажмите
 на
панели инструментов дизайнера
вычислительных схем. Подробнее см. документацию.
на
панели инструментов дизайнера
вычислительных схем. Подробнее см. документацию.
- Более тесная интеграция с дизайнером вычислительных схем: в частности, выходной файл или папка,
задаваемые в параметре созданного
элемента, автоматически отображаются в результатах запуска вычислительной схемы.

Помимо этого, в версии 33 появилась цветовая подсветка «Percentage Identity (colored)» для множественных выравниваний.
' в редакторе выравниваний")
В новую версию включены улучшения инфраструктуры для анализа данных высокопроизводительного секвенирования (NGS): интегрирован инструмент MetaPhlAn2, добавлены прочие мелкие улучшения.
Инструмент MetaPhlAn2 (METAgenomic PHyLogenetic ANalysis) предназначен для метагеномного профилирования данных полногеномного секвенирования. Вместе с инструментом поставляется база данных маркерных генов, выбранных из ~17,000 референсных геномов (из них ~13,500 геномы бактерий и архей, ~3,500 вирусов, ~110 эукариот).
В UGENE инструмент интегрирован в виде элемента вычислительных схем. Элемент включен в схему-пример «Parallel NGS reads classification»:

См. также:
- видео про метагеномные инструменты, появившиеся в версиях UGENE 1.31-1.32 (на английском языке);
- практическое занятие по метагеномике с использованием инструмента Kraken (на английском языке);
- если вы используете метагеномные инструменты в UGENE, пожалуйста, процитируйте данную публикацию.
В данный релиз включено исправление критической ошибки на выпущенной недавно операционной системе macOS Mojave.
Также исправлены ошибки:
- при экспорте консенсуса в редакторе данных секвенирования по Сэнгеру,
- при скачивании последовательности из базы данных Ensembl.
- Контроль качества NGS-данных: добавлен новый инструмент Trimmomatic, позволяющий обрезать
адаптерные последовательности Illumina,
обрезать концы прочтений по качеству, обрезать прочтения по длине и др.
 Инструмент встроен в виде элемента
вычислительных схем и
включен в несколько примеров схем (см. ниже, например).Для использования Trtimmomatic требуется
установить Java
Runtime Environment и сконфигурировать путь до «java» в настройках UGENE.
Инструмент встроен в виде элемента
вычислительных схем и
включен в несколько примеров схем (см. ниже, например).Для использования Trtimmomatic требуется
установить Java
Runtime Environment и сконфигурировать путь до «java» в настройках UGENE.
- Сборка de novo: инструмент SPAdes обновлен до версии 3.12.0. Графический интерфейс UGENE
позволяет теперь осуществлять сборку гибридных
данных от разных платформ секвенирования, например, сочетать при сборке прочтения Illumina и Oxford
Nanopore.
 Несколько примеров вычислительных схем с
инструментом SPAdes доступны из главного
меню программы «Tools > NGS data analysis > Reads de novo assembly (with SPAdes)».При
необходимости можно задать другие входные
данные для SPAdes. При этом должна быть указана хотя бы одна NGS-библиотека из следующих:
Несколько примеров вычислительных схем с
инструментом SPAdes доступны из главного
меню программы «Tools > NGS data analysis > Reads de novo assembly (with SPAdes)».При
необходимости можно задать другие входные
данные для SPAdes. При этом должна быть указана хотя бы одна NGS-библиотека из следующих:
- Парные (paired-end) / длинные парные (high-quality mate-pairs) / непарные прочтения Illumina или Ion Torrent
- Прочтения PacBio CCS
Дополнительно можно задать прочтения PacBio CLR, Oxford Nanopore, Sanger и/или контиги.
Подробнее см. параметр «Input data» у элемента SPAdes.
Инструмент SPAdes доступен на операционных системах macOS и Linux 64-бит.
- Метагеномика: добавлена новая инфраструктура для таксономической классификации NGS-данных,
включающая в себя такие инструменты, как
Kraken, CLARK, DIAMOND, WEVOTE и др. Для каждого инструмента предоставляются необходимые референсные
данные, например, таксономическая
информация с NCBI, RefSeq-геномы вирусов, бактерий, человека и др. Для установки/конфигурации
инструментов и данных рекомендуется использовать
онлайн-инсталлятор UGENE:
 Примеры
вычислительных схем доступны из главного меню «Tools > NGS data analysis > Metagenomics
classification». В частности, следующая
вычислительная
схема включает в себя
элементы Kraken, CLARK и DIAMOND. Еще один элемент WEVOTE объединяет результаты, полученные от этих
инструментов.
Примеры
вычислительных схем доступны из главного меню «Tools > NGS data analysis > Metagenomics
classification». В частности, следующая
вычислительная
схема включает в себя
элементы Kraken, CLARK и DIAMOND. Еще один элемент WEVOTE объединяет результаты, полученные от этих
инструментов. Помимо классификации, для каждого
инструмента встроен элемент, позволяющий
генерировать соответствующую базу данных (см., например, элемент «Build
Kraken Database«).Обратите внимание, что расчеты необходимо запускать на достаточно мощном
компьютере с оперативной памятью по
крайней мере 8 Гб. Пакет, включающий все данные, займет на диске примерно 230-240 Гб (подробнее см. данную
страницу). Т. к. в примеры вычислительных схем включены инструменты Trimmomatic и FastQC,
требуется установить на компьютере Java
Runtime Environment
и сконфигурировать путь до «java» в настройках UGENE. Инструменты доступны на операционных
системах macOS and Linux 64-бит.
Помимо классификации, для каждого
инструмента встроен элемент, позволяющий
генерировать соответствующую базу данных (см., например, элемент «Build
Kraken Database«).Обратите внимание, что расчеты необходимо запускать на достаточно мощном
компьютере с оперативной памятью по
крайней мере 8 Гб. Пакет, включающий все данные, займет на диске примерно 230-240 Гб (подробнее см. данную
страницу). Т. к. в примеры вычислительных схем включены инструменты Trimmomatic и FastQC,
требуется установить на компьютере Java
Runtime Environment
и сконфигурировать путь до «java» в настройках UGENE. Инструменты доступны на операционных
системах macOS and Linux 64-бит.
- Транскриптомика: добавлен новый инструмент StringTie для сборки транскриптов. Инструмент
интегрирован, как элемент
вычислительной схемы. Еще один элемент «StringTie
Gene Abundance Report» позволяет генерировать общий отчет для нескольких входных
NGS-библиотек.Следующая вычислительная схема доступна
из главного меню «Tools > NGS data analysis > RNA-Seq data analysis»:
 В данной схеме необходимо
создавать новый «Dataset» для
каждого образца (т. е., например, для пары FASTQ-файлов)!Обратите внимание, что инструменты StringTie и
TopHat доступны на операционных системах
macOS и Linux 64-бит. Для запуска инструментов Trimmomatic и FastQC tools необходимо установить Java
Runtime Environment и сконфигурировать путь до «java» в настройках UGENE.
В данной схеме необходимо
создавать новый «Dataset» для
каждого образца (т. е., например, для пары FASTQ-файлов)!Обратите внимание, что инструменты StringTie и
TopHat доступны на операционных системах
macOS и Linux 64-бит. Для запуска инструментов Trimmomatic и FastQC tools необходимо установить Java
Runtime Environment и сконфигурировать путь до «java» в настройках UGENE.
Также изменился подход к выделению аннотаций в редакторе последовательностей: для выделения аннотации вместе с соответствующим участком последовательности используйте двойной щелчок мыши. Одинарный щелчок мыши выделяет только аннотацию.
Помимо этого, рамка выделения стала заметнее:

- добавлен новый режим редактирования последовательности, cпособ изменения аннотаций при этом можно
настроить в диалоге, доступном из главного
меню Операции -> Редактирование -> Изменение аннотаций при редактировании
последовательности;

- добавлен новый режим отображения трансляций нуклеотидной последовательности;

- появилась возможность изменять размер выделенного участка в редакторе последовательности и редакторе
выравниваний;

- изменился вид по умолчанию детального представления последовательности: рамки считывания скрыты;
- стало возможным уменьшать вертикальный размер детального представление последовательности до высоты символов последовательности;
- исправлена подсветка символов расширенного алфавита в редакторе данных секвенирования по Сэнгеру;
- исправлены другие ошибки.
Подробнее про указанные функции можно прочитать в следующих главах документации (на английском языке):
- Editing Sequence
- Translating Nucleotide Sequence
- Selecting Sequence Region
- Selecting Alignment Region
См. также полный список изменений.
- доработка алгоритма выравнивания прочтений по Сэнгеру с учетом отзывов пользователей,

- исправление критической ошибки поиска по аминокислотным трансляциям нуклеотидной последовательности,
- исправление мелких ошибок и другие улучшения.
См. также полный список изменений.
- поддержка формата Vector NTI/AlignX,
- улучшение алгоритма выравнивания прочтений последовательностей по Сэнгеру,
- исправление ошибок, мелкие улучшения.
См. также полный список изменений.
- добавлен новый модуль для обработки данных секвенирования по Сэнгеру, включающий в себя функции:
- выравнивание прочтений на референсную последовательность,
- визуализация выровненных прочтений с хроматограммами,
- редактирование данных,
- экспорт результатов;
- исправление ошибок, мелкие улучшения.

См. также полный список изменений.
Новые возможности, улучшения, исправления
- Конструктор элементов для использования внешних инструментов в Дизайнере вычислительных схем
- Сборка длинных ридов ДНК с помощью CAP3
- Улучшенное определение форматов данных
- Просмотрищк BAM файлов: улучшенный расчет покрытия, интеграция со встроеными программами выравнивания
- Экспорт данных NGS: конвертация BAM в SAM
- Новый модуль: расчет флексибильности последовательностей ДНК
- Полный список доступен в баг трэкере.
Новые возможности, улучшения, исправления
- Новый модуль: редактор NGS данных Assembly Browser
- Отображение последовательностей: добавлена возможность по управлению количеством отображаемых аннотаций
- Круговой вид ДНК: добавлен удобный элемент управления — карта рестрикционных сайтов
- Модуль Phylip: добалена поддержка bootstrap для филогенетических деревьев
- Визуализатор 3D моделей: добавлена поддержка структурного выравнивания
- Добавлена автоматическая подсветка открытых рамок считывания
Полный список доступен в баг трэкере.
- Улучшена производительность UGENE Genome Aligner
- Добавлены новые элементы: запись и слияние аннотаций
- Конструктор Вычислительных Схем: запуск вычислительных схем прозводится в отдельных процессах
- Конструктор Вычислительных Схем: в поиска Смита-Ватермана добавлена поддержка файлов в формате FASTA в качестве поискового запроса
- Конструктор Вычислительных Схем: добален элемент HMM
- Конструктор Запросов: добавлена подержка конфигурации цепи ДНК (прямая или комплементарная) в запросах
- Конструктор Запросов: добавлены вомзожности по автоматичской подвсветке ферментов рестрикции.
- Исправлена ошибка с удаленными запросами к NCBI CDD
- Исправлена ошибка с экспортом проекта
- и другие исправления…
Полный список доступен в баг трэкере.
- Круговой вид: добавлено масштабирование
- Исправлен экспорт комплементарных аннотаций
- Редактор множественного выравнивания: исправления интерфейса
- Конструктор Вычислительных Схем: исправлены подсказки элементов
- Конструктор Вычислительных Схем: улучшения графического интерефейса
- Конструктор Вычислительных Схем: добавлен элемент для загрузки данных из удаленных БД
- Конструктор Запросов: добавлена поддержка направления стрэнда
- Поддрежан новый формат: pDRAW
- Добавлена возомжность экспотритровать группу аннотаци или всю таблицу в CSV
- Клонирование in silico: добавлена возможность редактирования фрагментов
- Клонирование in silico: аннотирование фрагментов в новой молекуле
- Клонирование in silico: исправлены ошибки конструирования новой молекулы
- и другие исправления…
Новые возможности, улучшения, исправление ошибок
- Клонирование in silico: разбивка на фрагменты и сшивка
- Новый инструмент: Конструктор запросов
- Поддержка внешних программ: BLAST+
- Поддержка внешних программ: TCoffee
- Добавлена поддержка OpenCL для Linux и Windows
- Улучшена система журналирования
- Добавлена система всплывающих напоминаний
- Несколько исправлений в экспорте аннотаций
- Исправлено поведение редактора последовательностей
- Улучшено распознавание кольцевых последовательностей
- Добавлен инструмент «Замена последовательности»
- Редактор множественного выравнивания: экспорт нуклеотидных выравниваний в аминокислотные
- Редактор множественного выравнивания: обратно-комплементированные последовательности
- Genome Aligner: сохранение индекса во внешнем файле
- Конструктор вычислительных схем: новый текстовый формат схем
- DotPlot: добавлен режимы увеличения
- DotPlot: улучшено выделение
- Улучшена обработка исключительных ситуаций
- Подсветка активного документа во вкладке проект
- Исправлен экспорт 3′-5′ аннотаций
- Исправлен отчёт о поиске аннотаций BLAST
- Исправлены проблемы в модуле поддержки внешних программ
- и многое другое…
Новые возможности и улучшения:
- Поддержка внешних программ: Blast;
- Поддержан новый формат: MEGA;
- Новая улучшенная система плагинов;
- Поддержка больших файлов( более 1 Гб) в сборке контигов.
- Улучшения инструментов командой строки: подробная система справки и диагностика ошибок;
- Исправлены падения во время построения молекулярных поверхностей для больших макромолекул;
- Множественные улучшения Конструктора вычислительных схем;
- Улучшена обработка исключительных ситуаций.
Новые возможности и улучшения:
- Запуск вычислительных задач на серверах Amazon EC2.
- Поддержка сторонних приложений: теперь в UGENE можно использовать инструменты множественного выравнивания Clustal and Mafft.
- Поддержка скриптинга для тонкой настройки сценариев Workflow Designer.
- Добавлена поддержка нового формата: Nexus
- Документация UGENE переработана и улучшена.
- Редактор множественного выравнивания: удаление столбцов с относительным числом пропусков;
- DotPlot: поиск обратных повтор, изменение масштаба, синхронизация с последовательностью;
- Переименование последовательностей, экспорт последовательностей с аннотациями;
Новые модули и возможности:
- Визуальный анализ гомологов — модуль DotPlot.
- Поддержка формата SAM, а также импорт значений качества PHRED для последовательностей ДНК;
- Продвинутый анализ хроматограм: обратные и компмлементарные хроматограмы, синхронное отображение;
- Новые опции инструментов для сборки контигов: BowTie и UGENE Genome Aligner;
- Пакетный режим работы для анализа последовательностей, основанного на весовых матрицах;
- Новые сценарии для дизайнера вычислительных схем, еще больше возможностей для использования UGENE как утилиты командой строки.
Новые модули и возможности:
- Переименование: congene -> ugene, ugene -> ugeneui
- Новый модуль: выравнивание геномов;
- Новый модуль: bowtie;
- Добавлен функционал сборки контигов;
- Добавлена поддержка частотных и весовых матриц;
- Редактор множественного выравнивания: добавлен новый режим увеличения;
- Улучшен модуль Circular view.
- К просмотровщику последовательностей добавлен просмотр панорамы;
- Интеграция с JASPAR и UniPROBE.
- Добавлено лого выравнивания.
- Улучшен консольный интерфейс.
- Конструктор схем: добавлена поддержка консольного режима.
- Переработан модуль удалённых запросов.
- В импорте CSV добавлен скриптинговый фреймворк.
- Переработан просмотровщик деревьев, добавлены новые режимы.
Новые модули и возможности:
- Новый модуль: Circular view;
- Модуль экспорта: добавлена функция обратной трансляции;
- Реализован функционал импорта аннотаций из CSV;
- MSA Editor: добавлен режим консенсуса, улучшен интерфейс;
- Улучшение просмотрщика филогенетических деревьев;
- Конструктор схем: добавлена возможность удалённого запуска заданий;
- Рестрикционные энзимы: теперь возможно создание пользовательских баз в формате BAIROCH;
- Доступен для скачивания бинарный дистрибутив для Linux;
- И многое другое…
Новые модули и возможности:
- KAlign: новый модуль для расчета множественного выравнивания, основанный на известном алгоритме; отличается высокой производительностью ( работает даже быстрее чем MUSCLE!) и аккуратностью;
- Поддержка нового формата: GFF;
- Улучшение просмотрщика филогенетических деревьев: поддержка формата Newick;
- Модуль Phylip: поддержка опций при расчете матрицы дистанций;
- Поддержка прокси с аутентификацией.
Исправления:
- Алгоритм Смита-Ватермана работает корректно при вызове из графической оболочки;
- В модуле MUSCLE улучшено управление памятью;
- Исправлена ошибка с некорректным удалением документов из Project View;
- Улучшено распознавание форматов;
- и многое другое…
Новые модули:
- Модуль Phylip для построения филогенетических деревьев. Основан на оригинальном пакете Phylip
3.6.
- Использует алгоритм Neighbor Joining для построения деревьев;
- Включены возможности по визуализации деревьев;
- Интеграция с редактором множественных выравниваний.
- Поддержка распределенных вычислений в UGENE
- Удаленное выполнение задач на сторонних комьютерах;
- Запуск HMMER3, Smith-Waterman, MUSCLE на удаленных машинах.
Улучшения:
- Поиск тандемных повторов;
- Поддержка переносимости файла проекта;
- Экспорт проекта;
- Интеграция с удаленными базами данных: NCBI, PDB, Swiss Prot;
- Визуализация биополимеров: режим стерео просмотра;
- и многое, многое другое…
Новые модули:
- MUSCLE 4 — интеграция новейшего инструмента для множественного выравнивания, автор которого — Robert C. Edgar;
- Адаптер CUDA — технический модуль, позволяющий использовать для вычислений видеокарты с поддержкой CUDA (только для Windows).
Новая функциональность и исправления:
- Пользователи Windows могут опробовать оптимизированный для CUDA алгоритм Смита-Ватермана;
- Добавлена одна из наиболее ожидаемых возможностей — редактирование последовательностей;
- Закончена интеграция пакета Primer 3 — теперь доступен полный набор настроек и параметров;
- Добавлена возможность экспорта аннотаций в CSV;
- Добавлены новые блоки в конструктор схем: «Запрос к удаленным БД» и «Извлечение аннотированных регионов»;
- Введена возможность сбора статистической информации об использовании UGENE (подробнее расскажет диалог, возникающий при первом запуске);
- HMM3 теперь поддерживает модели построенные с помощью HMM2;
- В модуль поиска энзимов рестрикции добавлен фильтр по числу результатов поиска;
- «Фантомные» документы теперь уничтожаются при закрытии проекта;
- В диалоге предсказания вторичной структуры исправлено неверное поведение селекторов региона;
- Исправлено некорректное поведение в параллельном режиме алгоритмов предсказания вторичной структуры, Primer3, парсера PDB-файлов;
- Серьезная ошибка затрагивающая пользователей Fedora Linux исправлена в алгоритме PSIPRED.
Новые модули:
- BALL — добавляет новые типы молекулярных поверхностей в модуль просмотра трехмерных моделей UGENE;
Новая функциональность и исправления:
- Добавлена возможность сохранения изображений множественного выравнивания и аннотированной последовательности (удобно для публикаций и печати);
- Исправлена критическая ошибка в congene;
- Исправлена критическая ошибка при попытке запуска HMM3 с моделями HMM2;
- Исправлена критическая ошибка при запуске нескольких задач Primer3 параллельно;
- Запрос к удаленным БД теперь стабилен на ОС Linux;
- Восстановлен пропавший алгоритм Смита-Ватермана;
- Задачи Primer3 теперь можно отменять;
- В модуль просмотра 3D-моделей добавлен выбор цвета выделения;
- Небольшое исправление внесено в отображение аннотированной последовательности;
Новые модули:
- HMM3 — новейший набор инструментов для работы с HMM-профайлами основанный на HMMER3;
- Primer3 — интегрированный инструмент Primer3 для подбора ПЦР-праймеров;
- GOR IV и PSIPRED — модули предсказания вторичной структуры белков;
- congene — консольная версия UGENE, позволяющая запускать вычислительные схемы из командной строки;
Новая функциональность и исправления:
- Добавлен расчет молекулярных поверхностей;
- Добавлена загрузка документов из баз данных PDB, Uniprot, Swissprot;
- Добавлена возможность создания нового документа;
- Улучшено редактирование аннотаций;
- Исправлены критические ошибки наведенные из Qt 4.5.0;
- В распараллеленный алгоритм MUSCLE внесены исправления, предотвращающие мертвую блокировку.
Улучшения и исправления ошибок:
- Теперь редактор множественного выравнивания поддерживает операции «Отменить» и «Повторить»;
- Исправлено поведение поиска по множественному выравниванию в случае разного регистра текста;
- Добавлены новые фильтры в поиск SITECON;
- Добавлена поддержка файлов CLUSTAL X;
- Алгоритм Смита-Ватермана теперь работает еще более точно;
- Исправлены ошибки параллельного вычисления в MUSCLE;
- Исправлены некоторые ошибки совместимости с Qt 4.5.0;
- А также множество других улучшений и исправлений.
Улучшения и исправления ошибок:
- Добавлена поддержка различных цветовых схем в редактор выравнивания;
- Добавлена блокировка камеры для нескольких трехмерных моделей;
- Просмотр последовательности оптимизирован для работы с гигантским количеством аннотаций (вплоть до 1 миллиона);
- Теперь аннотации могут быть подписаны как именем, так и значениями квалификаторов.
- Включен по умолчанию показ графиков качества хроматограммы для файлов ABIF;
- Исправлен ряд ошибок стабильности и совместимости в модулях BLAST, MUSCLE и HMMER;
- А также множество прочих улучшений и исправлений.
Новая функциональность:
- Новый модуль: «Smith-Waterman». Наиболее полная реализация алгоритма Смита-Ватермана выравнивания последовательностей;
- Новый модуль: «Restriction Enzymes». Маркирует ферменты рестрикции в последовательности. Поддерживает формат файлов базы данных REBASE;
- Новый модуль: «Repeat Finder». Ищет прямые и обратные повторы. Оптимизирован для быстрой работы с большими строками;
- Новый модуль: «DNA Stat». Удобные для экспорта и публикаций статистические отчеты;
- Конструктор схем: добавлена визуализация процесса исполнения схемы;
- Конструктор схем: добавлены примеры реальных схем;
- Конструктор схем: теперь можно изменять размер блоков, привязывать блоки к сетке, настраивать цвета и шрифты;
- Редактор выравнивания: добавлен показ смещений для каждой последовательности в выравнивании;
- Редактор выравнивания: добавлен поиск по выравниванию;
- Просмотр последовательности: добавлена блокировка масштаба и синхронизация между многими последовательностями;
- Просмотр последовательности: добавлена поддержка произвольного числа шкал обзора;
- Просмотр 3D: добавлен экспорт изображений в векторный формат (SVG);
- Просмотр 3D: добавлен поиск текущего файла по Web-ресурсам;
- Управление проектом: добавлены новые режимы отображения — группировка по типам объектов, выключение группировки;
- Управление проектом: для незагруженных документов показываются содержащиеся в них объекты;
- Производительность: теперь UGENE позволяет выбирать число используемых процессорных ядер, памяти и одновременных потоков;
- Модуль DNA Export: добавлен экспорт выравниваний в формат FASTA;
- Поддержаны новые форматы файлов: FASTQ, MMDB;
- Добавлена возможность индексирования больших файлов (> 10 Гб) для быстрого доступа к их содержимому;
- Добавлена поддержка произвольных схем оформления пользовательского интерфейса.
Улучшения и исправления ошибок:
- Модуль MUSCLE: одна из фаз алгоритма оптимизирована для работы на многоядерных процессорах;
- Модуль HMMER: внедрена оптимизация алгоритма использующая SSE2 инструкции;
- Модуль HMMER: hmmsearch оптимизирован для многоядерных процессоров (ускорение до 30 раз на четырехядерном процессоре с использованием SSE2);
- Модуль SITECON: >50 новых прокариотических профилей для поиска транскрипционных факторов сайтов связывания;
- Большинство диалогов и окон стали значительно удобнее и быстрее;
- Конструктор схем: теперь дополнительная информация о входных данных (ID в базе данных и т. д.) сохраняется на всех этапах вычислений.
- Конструктор схем: добавлено более интеллектуальное обращение с выходными данными — возможность перезаписывать либо добавлять информацию в конец файла.
- Управление проектом: добавлена возможность копирования и удаления документов;
- Создание аннотаций: теперь новые аннотации могут быть добавлены в новую таблицу аннотаций, которая будет автоматически загружена.
- Редактор выравнивания: добавлено изменение масштаба;
- Просмотр 3D: добавлено отображение нескольких моделей;
Улучшения и исправления ошибок:
- Исправлена наведенная из Qt 4.4 ошибка отображения трехмерных моделей на ОС Linux;
- Исправлена ошибка именования групп в случае вложенных групп;
- Форматы ABI и SCF теперь поддерживаются более полно;
- Отныне в именах аннотаций допускается использование символа ‘z’;
- Улучшена производительность отображения дерева аннотаций;
- Исправлен выбор региона для алгоритма SITECON;
- Исправлена критическая ошибка в конструкторе схем при запуске нескольких итераций;
- Теперь для коротких строк не содержащих символов ‘U’ и ‘T’ по умолчанию выбирается алфавит DNA вместо RNA;
- Теперь для форматов Genbank и FASTA допускаются символы табуляции;
- Бинарный пакет UGENE для дистрибутива Ubuntu разбит на две части в силу требований к пакетам попадающим в официальный репозиторий.
Улучшения и исправления ошибок:
- Исправлена ошибка не позволявшая открывать несколько окон на 64-битных системах;
- Исправлено аварийное завершение программы при открытии нескольких трехмерных моделей в одном окне;
- Исправлено аварийное завершение при открытии файла ABI с коротким именем сэмпла;
- Добавлен пункт меню «Проверить обновления»;
- Исправлено аварийное завершение при выборе комплементарного стренда для протеиновых последовательностей в диалоге «Поиск подстрок»;
- Исправлено ассоциирование объекта хроматограммы для форматов ABI и SCF;
- Исправлено автоопределение таблицы трансляции для формата PDB;
- Улучшен внешний вид надписей на ОС Linux и Mac OS;
- Улучшено поведение операции «Добавить объект в окно».
Новая функциональность:
- Поддержка ОС Linux. Выпущены пакеты для Ubuntu и Fedora;
- Новый модуль: Workflow Designer. Позволяет конструировать новые вычислительные процессы;
- Новый модуль: SITECON. Поиск сайтов связывания транскрипционных факторов;
- Новый модуль: BioStruct3D. Визуализация трехмерных моделей из файлов PDB;
- Поддержаны новые форматы: PDB и Stockholm;
- Добавлена поддержка множества сиквенсов в окне просмотра последовательности;
- Добавлена сортировка по значениям квалификаторов;
- Значения квалификаторов ‘db_xref’ сделаны ссылками на соответствующие базы данных;
- Теперь можно прятать транслированные и комплементарные строки;
- Поддержаны локализации — теперь в поставку UGENE входят русский и английский языки;
- Сделана поддержка отчетов о выполнении вычислительных задач;
Улучшения и исправления ошибок:
- Теперь в строке статуса показывается процент выполнения текущих задач;
- Исправлен ряд критических ошибок в модуле MUSCLE;
- Параметры по умолчанию модуля HMMER приведены в соответствие с параметрами оригинального HMMER;
- Отключены избыточные сообщения лога;
- Поддержан Drag&drop для всех типов файлов распознаваемых UGENE;
- >100 небольших исправлений и улучшений производительности и интерфейса.
Новая функциональность:
- Поддержка Mac OS 10.5 и 10.4;
- Создан редактор множественного выравнивания;
- Новый модуль: ORF Marker. Маркирует открытые рамки считывания;
- Новый модуль: ChromaView. Модуль добавляет возможность просмотра хроматограмм;
- Новый модуль: Remote request: Аннотирует выбранные регионы с помощью запросов к удаленным базам данных BLASTn, BLASTp и CDD. Может быть расширен для использования с другими сервисами с помощью скриптов;
- Новый модуль: UMUSCLE. Добавляет в систему алгоритм выравнивания MUSCLE.
- Внедрена прозрачная поддержка файлов на удаленных HTTP ресурсах;
- Внедрена поддержка произвольных скриптов для выполнения запросов подобных BLAST/CDD. Биндинги для всех GUI/Utility классов из Qt;
- Модуль DNAGraphPack поддерживает 4 новых типа графиков: GC & AT отклонения, Karlin Signature Difference и информационная энтропия;
- Теперь пользователю доступны для выбора любые таблицы трансляций;
- Добавлена поддержка формата SCF;
- Добавлена поддержка незагруженных документов, без удаления их из проекта.
Улучшения и исправления ошибок:
- При установке на Windows, UGENE ассоциирует с собой все поддерживаемые типы файлов;
- Улучшена интеграция модуля uHMMER с окном просмотра последовательности и редактором выравнивания;
- Модуль DNAGraphPack plugin now shows interval based graphs for large zooms to keep min/max information precise.
- Добавлена поддержка Drag&Drop между окном аннотаций и окном проекта;
- А также множество прочих улучшений и исправлений интерфейса, производительности и совместимости.
Новая функциональность:
- Поддержка форматов EMBL, ABI, CLUSTALW;
- Добавлены новые опции загрузки документов. Эту возможность удобно использовать для слияния нескольких последовательностей из входного файла в одну;
- Новый модуль: uHMMER. Интегрированные инструменты hmmsearch, hmmbuild и hmmcalibrate из пакета HMMER2.3.2;
- Новый модуль: DNA Export. Позволяет экспортировать выделенные либо аннотированные регионы в FASTA;
- Добавлена поддержка документов сжатых с помощь GZIP;
- Новый модуль: DNA Annotator. Позволяет производить поиск по аннотациям.
Улучшения и исправления ошибок:
- Добавлена возможность множественного выделения аннотаций в дереве;
- Теперь можно менять цвета, видимость и положение аннотаций;
- Переработан алгоритм поиска подстрок — исправлен ряд ошибок;
- Множество улучшений интерфейса и производительности.